
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Discrete polygonal supramolecular architectures of isocytosine-based Pt(II) complexes at the solution/graphite interface†
Mohamed
El Garah
a,
Stephan
Sinn
b,
Arezoo
Dianat
c,
Alejandro
Santana-Bonilla
c,
Rafael
Gutierrez
c,
Luisa
De Cola
b,
Gianaurelio
Cuniberti
 *cde,
Artur
Ciesielski
a and
Paolo
Samorì
*a
*cde,
Artur
Ciesielski
a and
Paolo
Samorì
*a
aLaboratoire de Nanochimie, ISIS & icFRC, Université de Strasbourg & CNRS, 8 allée Gaspard Monge, 67000 Strasbourg, France. E-mail: samori@unistra.fr
bLaboratoire de Chimie et des Biomatériaux Supramoléculaires, ISIS & icFRC, Université de Strasbourg & CNRS, 8 allée Gaspard Monge, 67000 Strasbourg, France
cInstitute for Materials Sciences and Max Bergmann Center of Biomaterials, TU Dresden, 01062 Dresden, Germany
dDresden Center for Computational Materials Science (DCCMS), TU Dresden, 01062 Dresden, Germany
eCenter for Advancing Electronics Dresden, TU Dresden, 01062 Dresden, Germany. E-mail: projects@tu-dresden.de
First published on 18th August 2016
Abstract
Polygonal supramolecular architectures of a Pt(II) complex including trimers, tetramers, pentamers and hexamers were self-assembled via hydrogen bonding between isocytosine moieties; their structure at the solid/liquid interface was unravelled by in situ scanning tunneling microscopy imaging. Density functional theory calculations provided in-depth insight into the thermodynamics of their formation by exploring the different energy contributions attributed to the molecular self-assembly and adsorption processes.
Molecular self-assembly at surfaces is a versatile approach to position functional groups with sub-nanometer precision over several hundreds of μm2 areas and thereby it allows the fine-tuning of numerous physico-chemical properties of the resulting nanostructures.1–6 The design of molecules comprising active groups represents a promising route for engineering functional surfaces and interfaces. Among various functional systems, luminescent Pt(II) complexes have attracted great interest during the last decade due to their appealing photophysical and redox properties, including strong absorption bands featuring onsets in the visible region as well as room temperature emission accompanied by a large Stokes shift.7 Neutral Pt(II) complexes bearing tridentate azolate-based ligands possess extraordinarily high phosphorescence quantum yields and long excited state lifetimes.8,9 Furthermore, due to their square-planar geometry such complexes display high propensity to undergo aggregation by means of the solvophobic effects, π–π and dispersive interactions. These properties can lead to supramolecular assemblies holding attractive features, because closed-shell metal–metal interactions of the Pt(II) centers strongly augment the emission properties.10–13
Scanning tunnelling microscopy (STM) is an extremely powerful tool to investigate supramolecular materials featuring atomic resolution. In particular, it is the method of choice for imaging structures and dynamics in molecular self-assembly on surfaces.14–18
Hydrogen bonding has been extensively used as a directional, non-covalent intermolecular interaction for designing and controlling the formation of complex supramolecular architectures. On solid surfaces, sophisticated structures can result from the formation of various hydrogen bonded patterns, including linear and cyclic motifs with different pairings such as O–H⋯O, N–H⋯O and N–H⋯N.19–23 Nucleobases have been previously reported to form cyclic quartet structures on the surface23–26 and pentamers in the solution.27 To the best of our knowledge, the formation of other types of cyclic motifs has not been discussed.
Here, we report on the self-assembly at the solid/liquid interface of Pt(II) complex Pt-Py-iCyt (Scheme 1) into discrete supramolecular architectures held together via intermolecular hydrogen bonds between isocytosine moieties. In particular, we show the formation of polycrystalline structures composed of various polygonal assemblies including trimers, tetramers, pentamers and hexamers stabilized by distinct hydrogen bond pairing.
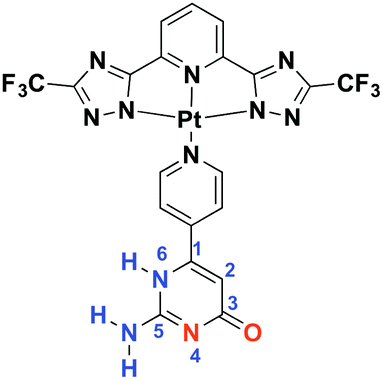 | ||
| Scheme 1 Chemical structure of the Pt(II) complex (Pt-Py-iCyt). Hydrogen-bonding donor and acceptor sites are indicated in blue and red, respectively. | ||
Supramolecular systems, where Pt(II) ions have been directly coordinated to nucleobases, have been the subject of numerous studies.28–31 Certain platinum complexes of purine and pyrimidine derivatives were found to present promising properties as antitumor drugs.32,33 Here we demonstrate for the first time that nucleobases, and in particular isocytosine, can be used to steer the 2D assembly of Pt(II) complexes through hydrogen bonding interactions between nucleobases. We support our experimental findings by performing density functional theory (DFT) calculations, and analysing both the strength of hydrogen bonding and the interactions with the graphite substrate. We also simulate the STM images to get an in depth understanding of the electronic features of the molecular architectures.
Initially, we investigated the self-assembly of Py-iCyt at the solid/liquid interface (see the ESI† for details). STM characterization revealed that Py-iCyt forms a supramolecular ribbon-like motif at the interface between the solution of Py-iCyt in 1-phenyloctane and highly oriented pyrolytic graphite (HOPG). Such a motif is in good agreement with previous observations on supramolecular structures of 6-methylisocytosine derivatives34,35 and 6-[4-(octyloxy)phenyl]isocytosine.36 This means that the functionalization of isocytosine with a pyridine unit does not influence its self-assembly behaviour at the solid/liquid interface.
We then extended our studies to the self-assembly of Pt-Py-iCyt at surfaces by applying a 4 μL drop of 100 ± 2 μM solution in 1-phenyloctane on a freshly cleaved HOPG substrate. The incorporation of the sterically demanding CF3-Pt moiety in the scaffold led to a dramatic change in the self-assembly behaviour. The STM image in Fig. 1a displays discrete supramolecular cyclic architectures. All these cyclic assemblies have been identified and are portrayed in Fig. 1b: trimers (iC3, white), tetramers (iC4, blue), pentamers (iC5, green), half of hexamers (1/2iC6, red) and hexamers (iC6, yellow).
 | ||
| Fig. 1 (a) Height STM image of Pt-Py-iCyt at the solution/HOPG interface. Scanning tunnelling parameters: average tunnelling current (It) = 25 pA, tip bias voltage (Vt) = −450 mV. (b) Model of different self-assembled molecular motifs. | ||
Statistical analysis of these experimentally observed cyclic arrangements was performed on ten independent STM experiments, also by exploring different regions of the samples. Fig. S9 in the ESI† summarizes the occurrence of each self-assembled discrete motif, which has been monitored and identified. Such an analysis revealed that two supramolecular species dominate, i.e. 1/2iC6 (45%) and iC6 (35%), suggesting that they are both energetically favoured over the other cyclic motifs, i.e. iC5, iC4 and iC3 assemblies. The analysis of the number of monomers involved in various motifs further supports such an observation. Fig. S9 (ESI†) shows that >80% of the adsorbed molecules is being involved in the formation of either iC6 (50%) or 1/2iC6 (32%) structures.
The stability of the different supramolecular assemblies and their corresponding electronic structures was computed at the density functional theory (DFT) level with the standard implementation in the CP2K package.
We also simulated the STM images of ordered molecular motifs (iC6, iC5, iC4, iC3 and 1/2iC6) and the results are presented in Fig. 2 (right panel). A good agreement is found between the experimental and simulated STM images when the complex is packed onto graphite in iC6, iC5, iC4, iC3 and 1/2iC6 (Fig. 2). All molecular motifs randomly co-exist on the HOPG surface. iC6 and 1/2iC6, which are formed, respectively, by six and three Pt-Py-iCyt, are stabilized by hydrogen bonding motifs of type O(3)⋯H–N(6) and N(4)⋯H–N(5), formed between isocytosine groups (Fig. 2). The self-assembly of the molecular architectures iC3, iC4 and iC5 can be explained by the formation of electrostatic interactions O(3)⋯H–N(6) occurring between the isocytosine groups of adjacent molecules. The different intermolecular interactions of the polygonal discrete architectures are explicitly illustrated in Fig. S15 in the ESI† as well as the self-assembly of the Py-iCyt ligand.
 | ||
| Fig. 2 (left) STM images of different Pt-Py-iCyt motifs. (middle) Simulated structures on graphene. (right) Simulated STM images of different motifs. | ||
The relevant electronic structural features of the molecular building block (Pt-Py-iCyt) are illustrated in Fig. S14 in the ESI.† Panels b–d show the HOMO−1, HOMO and LUMO of the complex. The gap between the HOMO and HOMO−1 states was computed to be 45 meV. The LUMO level of Pt-Py-iCyt is spread over the molecular skeleton whereas the HOMO−1 is located only on the fluorinated part (Pt-CF3). We also show the HOMO distribution of the different suggested molecular motifs as well as the ligand ribbons (Fig. S15, ESI†). The rather high localization degree of the HOMOs in the different motifs can be understood as follows: Pt has two free electrons, one in the d-level (5d9) and one free electron in the s level (6s2). Orbital localization is achieved by hybridization of the d free electron of Pt with the π orbitals generated by the ligand (four nitrogen atoms). Also, the four nitrogen atoms generate an effective crystal field splitting on Pt and therefore the d states jump to the upper part of the electronic structure and not the s state.
The interpretation of the molecular motifs monitored experimentally by STM is fully consistent with STM simulations. iC6 was generated through the formation of hydrogen bonds between isocytosine groups leading to a circular motif (Fig. 2). The observed contrast of different motifs – for example a dark center and a bright contour of iC6 (Fig. 2, right) – is due to HOMO levels of the assembled complexes. For this reason and because the STM images are recorded with a negative sample bias (occupied states), each individual Pt-Py-iCyt complex appears as a bright isolated protrusion in different molecular motifs. Three, four, five and six molecules are self-assembled to form, respectively, 1/2iC6 or iC3, iC4, iC5 and iC6 (details in the ESI†).
In order to investigate the mechanical stability of the different supramolecular motifs, we have calculated the dissociation energy for each suggested supramolecular structure shown in the middle panel of Fig. 2. We have defined the dissociation energy as:
| Ediss = EN − EN−1 − ESM | (1) |
Fig. 3 shows the computed dissociation energies of the molecular motifs. The number of hydrogen bonds, their nature and the degree of molecular packing play a crucial role in the dissociation energy of the molecular motifs. The highest dissociation energies were observed for iC6 and 1/2iC6 motifs, both formed through O(3)⋯H–N(6) and N(4)⋯H–N(5) hydrogen bonding. Lower Ediss values have been observed for iC3–iC5 structures, which are stabilized via a weaker type of H-bonding, i.e. O(3)⋯H–N(6). The higher degree of molecular packing from iC3 to iC5 leads to a slight decrease of dissociation energies as a result of the repulsive interaction between the fluorine groups.
 | ||
| Fig. 3 Calculated adsorption (Ead, red), dissociation (Ediss, green) and the total (Etot = Ead + Ediss, blue) energies of different molecular motifs self-assembled by Pt-Py-iCyt complex. | ||
The optimized structures of the different supramolecular motifs on the graphene surface are shown in Fig. 2 (middle panel). The adsorption energies Ead of these structures are reported in Fig. 3. Rather weak yet comparable interaction energies of Pt-Py-iCyt with the graphene substrate were found for all molecular structures (below 30 meV per atom). The STM analysis of films prepared from Pt-Py-iCyt solutions revealed the formation of polycrystalline structures dominated by iC6 and 1/2iC6 assemblies (ca. 80%; Fig. S9 in the ESI†), which is in good agreement with the results of the theoretical studies. In particular, both iC6 and 1/2iC6 assemblies are being favoured among all Pt-Py-iCyt structures, a finding, which can be directly correlated with the largest energies of dissociation. While the strength of intermolecular interactions plays a major role in the Pt-Py-iCyt self-assembly, the impact of the adsorption energies can be neglected, as Ead were found to be comparable among all Pt-Py-iCyt assemblies.
In summary, we have performed STM study and DFT analysis on the formation of different polygonal discrete cyclic supramolecular motifs formed by the isocytosine based Pt(II) complex at the solution/HOPG interface through hydrogen bonding interactions between isocytosine groups of Pt-Py-iCyt. Both the experimental and the theoretical results provided unambiguous evidence that the formation of hexameric and half-hexameric motifs of Pt-Py-iCyt molecules is thermodynamically favoured, as ruled by the strength of the association energy. Nevertheless, pentamers, tetramers and trimers are also observed at the solid/liquid interface. The approach described in this work can be exploited to control the supramolecular structure of Pt(II) complexes at the surface, and consequently as a way for tuning the photophysical properties of such materials.
This work was supported by the European Community through the project EC FP7 ICT-MOLARNET (318516) and the European Research Council project SUPRAFUNCTION (GA-257305), the Agence Nationale de la Recherche through the LabEx project Chemistry of Complex Systems (ANR-10-LABX-0026_CSC) and the International Center for Frontier Research in Chemistry. This work was also partly supported by the German Research Foundation (DFG) within the Cluster of Excellence “Center for Advancing Electronics Dresden”. Computational resources were provided by the ZIH at the Dresden University of Technology.
Notes and references
- J. V. Barth, G. Costantini and K. Kern, Nature, 2005, 437, 671–679 CrossRef CAS PubMed.
- M. Stöhr, M. Wahl, C. H. Galka, T. Riehm, T. A. Jung and L. H. Gade, Angew. Chem., Int. Ed., 2005, 44, 7394–7398 CrossRef PubMed.
- J. A. A. W. Elemans, R. Van Hameren, R. J. M. Nolte and A. E. Rowan, Adv. Mater., 2006, 18, 1251–1266 CrossRef CAS.
- G. P. Spada, S. Lena, S. Masiero, S. Pieraccini, M. Surin and P. Samorì, Adv. Mater., 2008, 20, 2433–2438 CrossRef CAS.
- T. Kudernac, S. B. Lei, J. A. A. W. Elemans and S. De Feyter, Chem. Soc. Rev., 2009, 38, 402–421 RSC.
- D. Bonifazi, S. Mohnani and A. Llanes-Pallas, Chem. – Eur. J., 2009, 15, 7004–7025 CrossRef CAS PubMed.
- J. A. G. Williams, Photochemistry and Photophysics of Coordination Compounds II, Springer, 2007, pp. 205–268 Search PubMed.
- C. A. Strassert, C. H. Chien, M. D. Galvez Lopez, D. Kourkoulos, D. Hertel, K. Meerholz and L. De Cola, Angew. Chem., Int. Ed., 2011, 50, 946–950 CrossRef CAS PubMed.
- N. K. Allampally, C. A. Strassert and L. De Cola, Dalton Trans., 2012, 41, 13132–13137 RSC.
- V. M. Miskowski and V. H. Houlding, Inorg. Chem., 1989, 28, 1529–1533 CrossRef CAS.
- B. Ma, J. Li, P. I. Djurovich, M. Yousufuddin, R. Bau and M. E. Thompson, J. Am. Chem. Soc., 2005, 127, 28–29 CrossRef CAS PubMed.
- M. Mydlak, M. Mauro, F. Polo, M. Felicetti, J. Leonhardt, G. Diener, L. De Cola and C. A. Strassert, Chem. Mater., 2011, 23, 3659–3667 CrossRef CAS.
- M. Mauro, A. Aliprandi, D. Septiadi, N. S. Kehr and L. De Cola, Chem. Soc. Rev., 2014, 43, 4144–4166 RSC.
- F. Rosei, M. Schunack, Y. Naitoh, P. Jiang, A. Gourdon, E. Laegsgaard, I. Stensgaard, C. Joachim and F. Besenbacher, Prog. Surf. Sci., 2003, 71, 95–146 CrossRef CAS.
- A. Nickel, R. Ohmann, J. Meyer, M. Grisolia, C. Joachim, F. Moresco and G. Cuniberti, ACS Nano, 2013, 7, 191–197 CrossRef CAS PubMed.
- D. den Boer, M. Li, T. Habets, P. Iavicoli, A. E. Rowan, R. J. M. Nolte, S. Speller, D. B. Amabilino, S. De Feyter and J. A. A. W. Elemans, Nat. Chem., 2013, 5, 621–627 CrossRef CAS PubMed.
- S. Haq, F. Hanke, J. Sharp, M. Persson, D. B. Amabilino and R. Raval, ACS Nano, 2014, 8, 8856–8870 CrossRef CAS PubMed.
- Y. Yu, J. Lin, Y. Wang, Q. Zeng and S. B. Lei, Chem. Commun., 2016, 52, 6609–6612 RSC.
- M. Lackinger, S. Griessl, W. M. Heckl, M. Hietschold and G. W. Flynn, Langmuir, 2005, 21, 4984–4988 CrossRef CAS PubMed.
- A. Llanes-Pallas, C.-A. Palma, L. Piot, A. Belbakra, A. Listorti, M. Prato, P. Samorì, N. Armaroli and D. Bonifazi, J. Am. Chem. Soc., 2009, 131, 509–520 CrossRef CAS PubMed.
- J. M. MacLeod, O. Ivasenko, C. Fu, T. Taerum, F. Rosei and D. F. Perepichka, J. Am. Chem. Soc., 2009, 131, 16844–16850 CrossRef CAS PubMed.
- O. Ivasenko and D. F. Perepichka, Chem. Soc. Rev., 2011, 40, 191–206 RSC.
- M. El Garah, R. C. Perone, A. S. Bonilla, S. Haar, M. Campitiello, R. Gutierrez, G. Cuniberti, S. Masiero, A. Ciesielski and P. Samorì, Chem. Commun., 2015, 51, 11677–11680 RSC.
- R. Otero, M. Schock, L. M. Molina, E. Laegsgaard, I. Stensgaard, B. Hammer and F. Besenbacher, Angew. Chem., Int. Ed., 2005, 44, 2270–2275 CrossRef CAS PubMed.
- W. Xu, R. E. A. Kelly, H. Gersen, E. Laegsgaard, I. Stensgaard, L. N. Kantorovich and F. Besenbacher, Small, 2009, 5, 1952–1956 CrossRef CAS PubMed.
- L. K. Wang, H. H. Kong, C. Zhang, Q. Sun, L. L. Cai, Q. G. Tan, F. Besenbacher and W. Xu, ACS Nano, 2014, 8, 11799–11805 CrossRef CAS PubMed.
- M. Cai, A. L. Marlow, J. C. Fettinger, D. Fabris, T. J. Haverlock, B. A. Moyer and J. T. Davis, Angew. Chem., Int. Ed., 2000, 39, 1283–1285 CrossRef CAS.
- B. T. Khan and S. M. Zakeeruddin, Transition Met. Chem., 1991, 16, 119–121 CrossRef CAS.
- H. Rauter, I. Mutikainen, M. Blomberg, C. J. L. Lock, P. AmoOchoa, E. Freisinger, L. Randaccio, E. Zangrando, E. Chiarparin and B. Lippert, Angew. Chem., Int. Ed., 1997, 36, 1296–1301 CrossRef CAS.
- D. Gupta, M. Huelsekopf, M. M. Cerda, R. Ludwig and B. Lippert, Inorg. Chem., 2004, 43, 3386–3393 CrossRef CAS PubMed.
- D. Gupta, M. Roitzsch and B. Lippert, Chem. – Eur. J., 2005, 11, 6643–6652 CrossRef CAS PubMed.
- A. G. Quiroga, J. Inorg. Biochem., 2012, 114, 106–112 CrossRef CAS PubMed.
- X. Wang, X. Wang and Z. Guo, Acc. Chem. Res., 2015, 48, 2622–2631 CrossRef CAS PubMed.
- L. M. Toledo, K. Musa, J. W. Lauher and F. W. Fowler, Chem. Mater., 1995, 7, 1639–1647 CrossRef CAS.
- P. R. Lowe, C. H. Schwalbe and G. J. B. Williams, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1987, 43, 330–333 CrossRef.
- A. Ciesielski, S. Colella, L. Zalewski, B. Bruchmann and P. Samorì, CrystEngComm, 2011, 13, 5535–5537 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, synthesis, and details of DFT calculations. See DOI: 10.1039/c6cc05087e |
| This journal is © The Royal Society of Chemistry 2016 |