
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A traceless photocleavable linker for the automated glycan assembly of carbohydrates with free reducing ends†
M.
Wilsdorf
a,
D.
Schmidt
ab,
M. P.
Bartetzko
ab,
P.
Dallabernardina
ab,
F.
Schuhmacher
ab,
P. H.
Seeberger
ab and
F.
Pfrengle
*ab
aDepartment of Biomolecular Systems, Max-Planck-Institute of Colloids and Interfaces, Am Mühlenberg 1, 14476 Potsdam, Germany. E-mail: Fabian.Pfrengle@mpikg.mpg.de
bFreie Universität Berlin, Institute of Chemistry and Biochemistry, Arnimallee 22, 14195 Berlin, Germany
First published on 15th July 2016
Abstract
We report a traceless photocleavable linker for the automated glycan assembly of carbohydrates with free reducing ends. The reductive-labile functionality in the linker tolerates all commonly used reagents and protocols for automated glycan assembly, as demonstrated with the successful preparation of nine plant cell wall-related oligosaccharides, and is cleaved by hydrogenolysis.
Carbohydrates play a central role in materials science, biology and medicine.1 In order to study the structure–activity relationship of glycans, homogeneous and pure samples which are obtained through chemical synthesis are indispensable. Automated glycan assembly has evolved as a platform to rapidly access these complex glycans.2 The automated process assembles a diverse set of monosaccharide building blocks into oligosaccharide products without the need for manual intervention. Starting from a resin-bound linker, iterative glycosylation and deprotection steps systematically elongate the glycan chain. Photolabile linker 1 (cf.Fig. 1) is often employed to link the growing glycan to the solid support.3 This linker is cleaved in a continuous flow photoreactor followed by global deprotection to release glycans containing an aminoalkyl-linker at the reducing end. The amino group of the alkyl linker is utilized to attach glycans to microarray surfaces4 and carrier proteins required in the study and development of conjugate vaccine candidates.5
 | ||
| Fig. 1 Photocleavable linker 1 and traceless linker 2 for the automated assembly of glycans with an aminoalkyl linker and a free reducing end, respectively. | ||
Naturally occurring free reducing glycans are also highly desirable, since synthetic samples are frequently needed as standards for LC/MS measurements6 or biological assays that do not tolerate the presence of unnatural modifications. To access glycans with free reducing ends by solid-phase chemistry, base-labile linker systems have been used that incorporate a benzylic group to release the anomeric hydroxyl after a final hydrogenation step.7 Based on this precedence, we envisioned that the introduction of a benzyl moiety into photocleavable linker 1 would allow for rapid access to free reducing glycans after photo-induced cleavage of the glycans from the solid support followed by hydrogenolysis. Here, we report the synthesis and use of traceless linker 2, whose applicability was demonstrated in the automated assembly of various complex glycans with free reducing ends (Fig. 1).
Following the established route towards photo-cleavable linker 1, the preparation of 2 required only the substitution of the 5-aminopentanol portion for benzylic aminoalcohol 5, which is commercially available but very expensive. We designed a short and scalable route towards aminoalcohol 5 that provides an attractive alternative to the available literature procedures.8 Readily available terephthaldehyde (3) was selectively reduced once with NaBH4 and condensed with nitromethane in an Henry reaction to form nitroalkene 4.9,10 Subsequent conjugate reduction of the nitroalkene with NaBH4 in DMSO gave the corresponding nitroalkane (not shown) which was further reduced to the primary amine by hydrogenation and then isolated as its hydrochloride salt 5. With sufficient quantities of the aminoalcohol in hand, photocleavable linker 7 was obtained by reductive amination of 5 with aryl aldehyde 6 followed by protection of the resulting secondary amine with CbzCl.3 This short route was readily scaled up for the preparation of a seven gram batch of the product. Subsequent functionalization of Merrifield resin by alkylation with four equivalents of linker 7 finally provided resin-bound linker 2 with a loading determined to be approximately 0.26 mmol g−1 (Scheme 1).3,11 Excess linker 7 was efficiently recovered after functionalization.
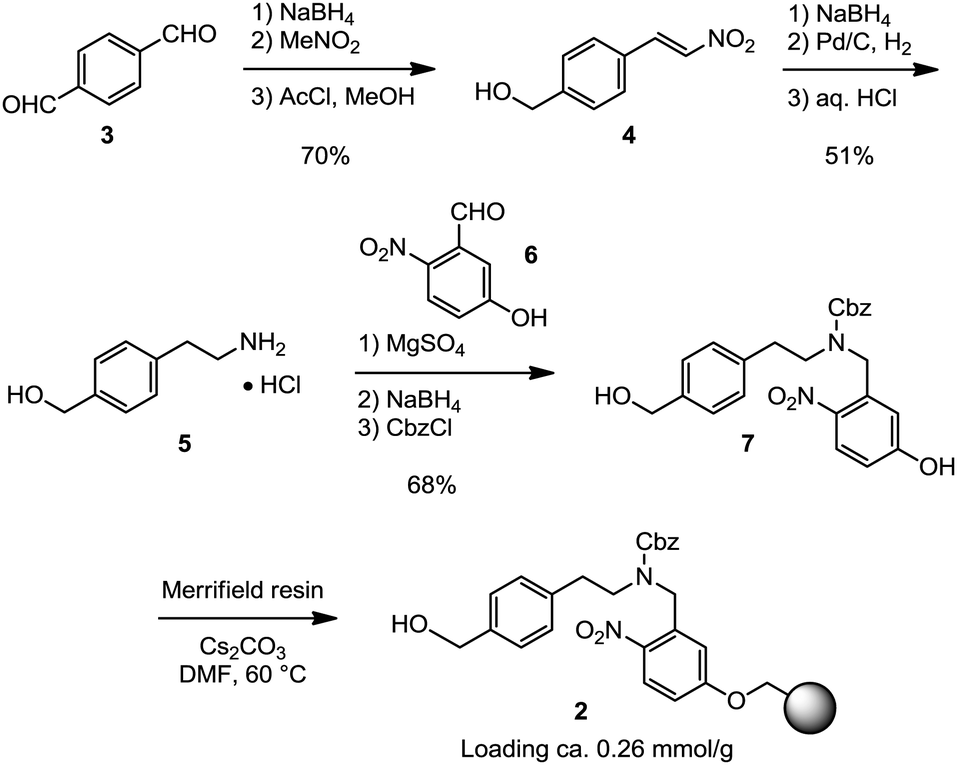 | ||
| Scheme 1 Gram-scale preparation of the resin-bound linker 2. | ||
The automated assembly of fully-protected cellulose tetrasaccharide 9 was selected as a proof-of-principle study for the utility of the new linker (Scheme 2). The synthesis of 9 included four iterative cycles of glycosylation and Fmoc-deprotection steps employing monosaccharide building block 8, followed by photolytic cleavage of the linker from the solid-support prior to final HPLC purification. Target molecule 9 was isolated in 67% yield and thus virtually with the same efficiency as aminopentyl-substituted cellulose derivative 10 that was obtained using linker 1. Notably, the presence of an additional benzene moiety in the linker did not affect the efficiency of the glycosylation and deprotection steps in the automated assembly as well as the subsequent photolytical cleavage. Next, cellulose tetrasaccharide 9 was sequentially deprotected by methanolysis of the benzoate esters (with NaOMe) followed by hydrogenolysis of the benzyl ethers and the linker (H2, Pd/C), affording cellulose tetrasaccharide 11 as a mixture of α/β-anomers. During concentration of the crude product, the open chain form of 11 reacted partially with the primary amine 12 to form a hemiaminal ether.12 Acidic hydrolysis converts this mixture to the final product 11 before it was subjected to final HPLC purification. As verified by analytical HPLC, MS and 2D NMR spectroscopy, the tetrasaccharide product was obtained in high purity, albeit in a rather low yield (20%) over the two deprotection steps. The moderate yield may be attributed to the low solubility of the partially protected intermediate (not shown), impeding purification by reversed phase-HPLC. The deprotection sequence is currently optimized to increase the overall efficiency of the process (Scheme 3).
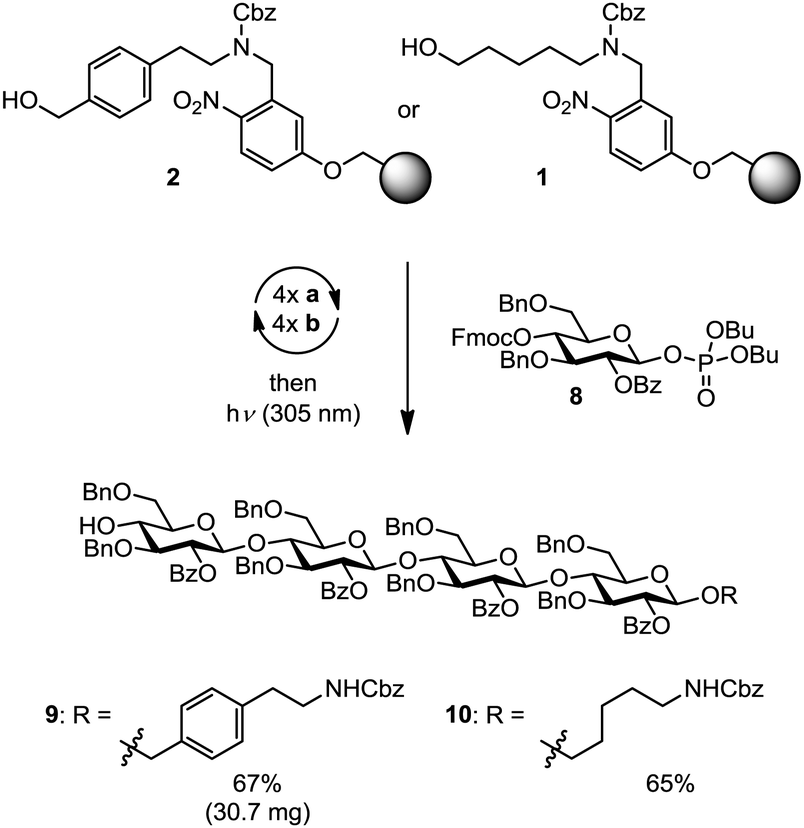 | ||
| Scheme 2 Head-to-head comparison of the traceless linker 2 and the aminoalkyl linker 1. Glycosylation step (a): 3.7 equiv. of 8, TMSOTf, CH2Cl2, −35 °C → −15 °C; deprotection step (b): 20% NEt3 in DMF, 25 °C. Yields are based on resin loading. | ||
 | ||
| Scheme 3 Global deprotection of cellulose tetrasaccharide 9. | ||
The utility of linker 2 was further illustrated by preparing a diverse set of plant cell wall-related oligosaccharides (Scheme 4). Seven orthogonally protected monosaccharide building blocks (BB) 8 and 13–18 were used for the automated glycan assembly of cellulose-, arabinoxylan-, and arabinogalactan-type oligosaccharides.13 Following iterative glycosylation and chemoselective deprotection steps, various linear and branched oligosaccharides up to hexasaccharides were obtained after photolytical cleavage from the solid support. These protected products were then subjected to methanolysis and hydrogenation to afford pure glycans 19–26 in 3–18% overall yield. For the automated assembly of the xylan oligosaccharides 20–23, the use of only 2.8 equiv. of building block per glycosylation step was sufficient, whereas for the construction of the notoriously difficult to synthesize Gal-β-1,3-Gal-linkage (24–26) eight equiv. of building block were necessary.13b Linker 2 proved to be compatible with all common protocols for automated glycan assembly, including glycosylations with glycosyl phosphates and thioglycosides, the removal of protecting groups such as Fmoc, Lev and Nap, as well as the capping of free hydroxyl groups as benzoyl esters.
 | ||
Scheme 4 Automated assembly of plant cell wall oligosaccharides 11, 19–26. Synthesizer modules (a): one or two cycles of 1.4–4.0 equiv. of BB, TMSOTf, CH2Cl2, −35 °C or −30 °C (5 min) → −15 °C or −20 °C (30 min); (b): two cycles of 1.4–4.0 equiv. of BB, NIS/TfOH, CH2Cl2/dioxane (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), −40 °C (5 min) → −20 °C (40 min); (c): 3 cycles of 20% Et3N in DMF, 25 °C (5 min); (d): 3 cycles of N2H4·HOAc (0.15 M) in pyridine/AcOH/H2O (4:1:0.25), 25 °C (30 min); (e): six cycles of DDQ (0.10 M) in DCE/MeOH/H2O (64:16:1), 40 °C (20 min); (f): three cycles of Bz2O (0.50 M in DCE) and DMAP (0.25 M in DCE), pyridine, 40 °C (30 min). Photolytical cleavage (g): hν (305 nm), DCM, rt. Global deprotection (h): NaOMe, MeOH, rt (16 h); (i): H2, 10% Pd/C, EtOAc/MeOH/H2O/AcOH, rt (16 h). Yields are based on resin loading. The letter codes below the structures represent the synthesizer modules and deprotection steps applied in the respective syntheses. :1), −40 °C (5 min) → −20 °C (40 min); (c): 3 cycles of 20% Et3N in DMF, 25 °C (5 min); (d): 3 cycles of N2H4·HOAc (0.15 M) in pyridine/AcOH/H2O (4:1:0.25), 25 °C (30 min); (e): six cycles of DDQ (0.10 M) in DCE/MeOH/H2O (64:16:1), 40 °C (20 min); (f): three cycles of Bz2O (0.50 M in DCE) and DMAP (0.25 M in DCE), pyridine, 40 °C (30 min). Photolytical cleavage (g): hν (305 nm), DCM, rt. Global deprotection (h): NaOMe, MeOH, rt (16 h); (i): H2, 10% Pd/C, EtOAc/MeOH/H2O/AcOH, rt (16 h). Yields are based on resin loading. The letter codes below the structures represent the synthesizer modules and deprotection steps applied in the respective syntheses. | ||
In conclusion, we have synthesized nine plant cell wall-related glycans using a novel traceless photocleavable linker that enables the automated glycan assembly of carbohydrates with free reducing ends. The linker complements the photocleavable linker for the synthesis of aminoalkyl-functionalized glycans and provides the oligosaccharides in comparable yields. The opportunity to produce large sets of related glycans with free reducing ends holds great potential for the preparation of standards for LC/MS- and ion mobility mass spectrometry experiments.6,14
We gratefully acknowledge financial support from the Max Planck Society, the German Research Foundation (DFG, Emmy Noether program PF850/1-1 to FP, fellowship Wi4356/2-1 to MW, and the SFB765), the Fonds der Chemischen Industrie (Liebig-fellowship to FP) and an ERC Advanced Grant (AUTOHEPARIN to PHS).
Notes and references
- (a) N. Taniguchi, T. Endo, G. W. Hart, P. H. Seeberger and C.-H. Wong, Glycoscience: Biology and Medicine, Springer, Tokyo, 2015 Search PubMed; (b) S. Dumitriu and V. Popa, Polymeric Biomaterials: Structure and Function, CRC Press, Boca Raton, 2013 Search PubMed.
- (a) O. J. Plante, E. R. Palmacci and P. H. Seeberger, Science, 2001, 291, 1523–1527 CrossRef CAS PubMed; (b) L. Kröck, D. Esposito, B. Castagner, C.-C. Wang, P. Bindschädler and P. H. Seeberger, Chem. Sci., 2012, 3, 1617–1622 RSC; (c) P. H. Seeberger, Acc. Chem. Res., 2015, 48, 1450–1463 CrossRef CAS PubMed.
- (a) S. Eller, M. Collot, J. Yin, H. S. Hahm and P. H. Seeberger, Angew. Chem., Int. Ed., 2013, 52, 5858–5861 CrossRef CAS PubMed; (b) O. Calin, S. Eller and P. H. Seeberger, Angew. Chem., Int. Ed., 2013, 52, 5862–5865 CrossRef CAS PubMed; (c) J. Kandasamy, F. Schuhmacher, H. S. Hahm, J. C. Klein and P. H. Seeberger, Chem. Commun., 2014, 50, 1875–1877 RSC; (d) R. J. Fair, H. S. Hahm and P. H. Seeberger, Chem. Commun., 2015, 51, 6183–6185 RSC.
- C. R. Rillahan and J. C. Paulson, Annu. Rev. Biochem., 2011, 80, 797–823 CrossRef CAS PubMed.
- (a) Z. Guo and G.-J. Boons, Carbohydrate-Based Vaccines and Immunotherapies, John Wiley & Sons, New Jersey, 2009 Search PubMed; (b) F. Broecker, C. Anish and P. H. Seeberger, Methods Mol. Biol., 2015, 1331, 57–80 CrossRef PubMed.
- M. P. Campbell, T. Nguyen-Khuong, C. A. Hayes, S. A. Flowers, K. Alagesan, D. Kolarich, N. H. Packer and N. G. Karlsson, Biochim. Biophys. Acta, 2014, 1844, 108–116 CrossRef CAS PubMed.
- (a) X. Wu, M. Grathwohl and R. R. Schmidt, Angew. Chem., Int. Ed., 2002, 41, 4489–4493 CrossRef CAS; (b) K. Tanaka, Y. Fujii, H. Tokimoto, Y. Mori, S.-I. Tanaka, G.-M. Bao, E. R. O. Siwu, A. Nakayabu and K. Fukase, Chem. – Asian J., 2009, 4, 574–580 CrossRef CAS PubMed; (c) T. J. Boltje, J.-H. Kim, J. Park and G.-J. Boons, Nat. Chem., 2010, 2, 552–557 CrossRef CAS PubMed.
- (a) L. Pignataro, S. Carboni, M. Civera, R. Colombo, U. Piarulli and C. Gennari, Angew. Chem., Int. Ed., 2010, 49, 6633–6637 CrossRef CAS PubMed; (b) J.-P. Behr and J.-M. Lehn, Helv. Chim. Acta, 1980, 63, 2112–2118 CrossRef CAS.
- (a) J. M. Lopchuk, R. P. Hughes and G. W. Gribble, Org. Lett., 2013, 15, 5218–5221 CrossRef CAS PubMed; (b) S. Peterli, D. Hubmann, U. Sequin, H. Mett and P. Traxler, Helv. Chim. Acta, 1994, 77, 59–69 CrossRef CAS.
- The generation of partially O-acetylated nitroalkene required a subsequent acidic methanolysis to afford pure 4.
- For loading determination, see the ESI†.
- W. Pigman, E. A. Cleveland, D. H. Couch and J. H. Cleveland, J. Am. Chem. Soc., 1951, 73, 1976–1979 CrossRef CAS.
- (a) D. Schmidt, F. Schuhmacher, A. Geissner, P. H. Seeberger and F. Pfrengle, Chem. – Eur. J., 2015, 21, 5709–5713 CrossRef CAS PubMed; (b) M. Bartetzko, F. Schuhmacher, H. S. Hahm, P. H. Seeberger and F. Pfrengle, Org. Lett., 2015, 17, 4344–4347 CrossRef CAS PubMed; (c) P. Dallabernardina, F. Schuhmacher, P. H. Seeberger and F. Pfrengle, Org. Biomol. Chem., 2016, 14, 309–313 RSC.
- J. Hofmann, H. S. Hahm, P. H. Seeberger and K. Pagel, Nature, 2015, 526, 241–244 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures for solution-phase reactions and automated glycan assembly and characterization data for all compounds. See DOI: 10.1039/c6cc04954k |
| This journal is © The Royal Society of Chemistry 2016 |