
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnion recognition by a bidentate chalcogen bond donor†
Graham E.
Garrett
,
Elisa I.
Carrera
,
Dwight S.
Seferos
* and
Mark S.
Taylor
*
Department of Chemistry, University of Toronto, Toronto, ON M5S 3H6, Canada. E-mail: mtaylor@chem.utoronto.ca; dseferos@chem.utoronto.ca
First published on 4th July 2016
Abstract
Perfluoroaryl-substituted tellurophenes act as anion receptors through noncovalent chalcogen bonding interactions. Linking two tellurophenes through an ethynylene group results in a significant level of chelate cooperativity, thus demonstrating that chalcogen bonding can be used to achieve multidentate anion recognition.
Chalcogen bonding is the noncovalent interaction that occurs when an electron-deficient, covalently bonded group 16 element (the chalcogen bond donor) is attracted to a Lewis basic site (the chalcogen bond acceptor, Fig. 1). The attractive nature of this interaction, arising from a combination of electrostatic, dispersion and charge transfer components, is supported by computational studies.1 These calculations have revealed similarities between chalcogen bonding and halogen bonding, the interaction of Lewis bases with group 17 elements.2 Both interactions are most favourable for the heaviest elements in the group (Te > Se > S > O and I > Br > Cl > F), and display directionality, with the preferred geometry having a 180° Y⋯E–R or Y⋯X–R angle (where Y is the Lewis base, E is a chalcogen, X is a halogen and R is a generic substituent). X-ray crystallography has provided substantial evidence that sulfur, selenium and tellurium compounds engage in attractive, directional interactions with Lewis bases in the solid state.3,4 Self-association of chalcogen-based heterocycles (e.g., chalcogenadiazoles, chalcogenazoles) into chains, ribbons or cyclic aggregates is a notable manifestation of this effect.5–7 A recent study has shown that isotellurazole-N-oxides form cyclic oligomers (e.g., tetramers, hexamers) that persist in solution and in the gas phase.8
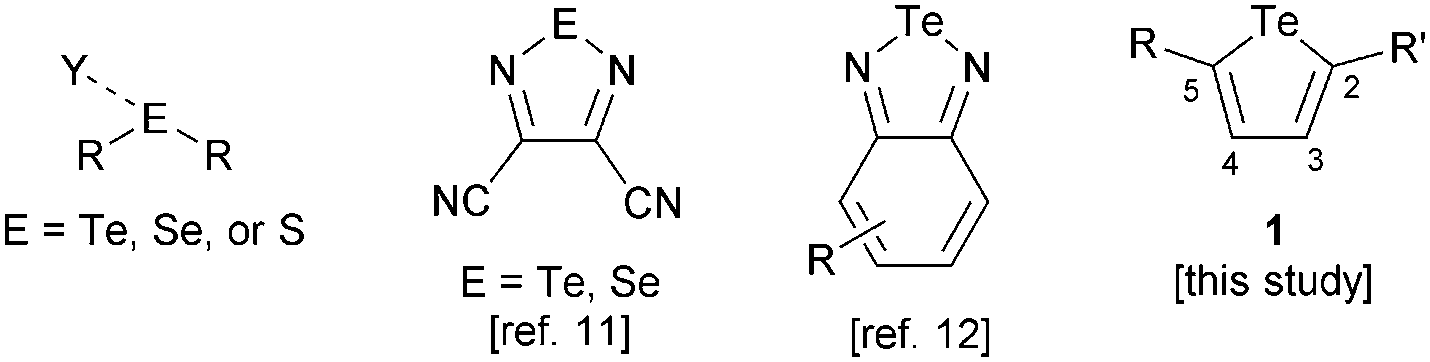 | ||
| Fig. 1 General depiction of a chalcogen bonding interaction; structures of the (benzo)chalcogenadiazole donors employed in previous studies, and the tellurophenes explored in the present work. | ||
Whereas considerable progress towards understanding and applying halogen bonding in the solution phase has been achieved over the past decade,9 only limited quantitative data are available regarding chalcogen bonding in solution. A telluronium substituent increased the fluoride affinity of a boron-based receptor through an attractive R3Te+⋯F− interaction.10 Zibarev and co-workers used optical absorbance spectroscopy to determine association constants of dicyanotelluradiazole and dicyanoselenadiazole with I− and PhS−, respectively, in organic solvents (Fig. 1).11 We carried out a systematic study of the interactions of anions with benzotelluradiazoles, showing how the strengths of these chalcogen bonds vary upon changing the anionic component, solvent and benzotelluradiazole substitution pattern.12 Bonding of the chalcogen element to a relatively electronegative imino group gives rise to a region of positive electrostatic potential (σ-hole13) that contributes to the donor ability of these chalcogenadiazole derivatives. However, we found that the Te–N bonds of benzotelluradiazoles are sites of reactivity, resulting in incompatibility with protic solvents and certain nucleophilic species. Donors having chalcogen–carbon bonds should possess advantages in terms of chemical stability, and the ability to incorporate substituents at C2 and C5 provides a way to exert steric and/or electronic effects on the tellurium center or to introduce additional functional groups (e.g., for the construction of multidentate receptors). The present study is aimed at probing the chalcogen bond donor ability of tellurophene derivatives (e.g., 1, Fig. 1). We show that an electron-deficient 2,5-diaryltellurophene displays appreciable anion affinity in organic solvent. A significant increase in affinity is gained using a bis(tellurophene) receptor in which the chalcogen-based heterocycles are joined by an ethynylene linker, thus demonstrating that bidentate chalcogen bonding can be used to achieve anion recognition.
We began by evaluating 2,5-diaryltellurophenes 1a–1d as monodentate chalcogen bond donors (Fig. 2). Derivatives having at least one electron-withdrawing arene substituent were selected, in keeping with the envisioned role of the tellurophene as a Lewis acid in this interaction. The compounds were prepared either by reaction of the conjugated diyne with sodium telluride14 or by palladium ipso-arylation of 2,5-bis[(diphenyl)hydroxymethyl]tellurophene,15 as described previously.16 Anion affinities were assessed by monitoring changes in optical absorbance spectra upon addition of tetrabutylammonium chloride (Bu4N+Cl−) in tetrahydrofuran (THF) at 298 K. The bis(4-cyanophenyl)- and bis(4-(trifluoromethyl)phenyl)-substituted congeners 1a and 1b, as well as unsymmetrically substituted 1c, did not interact with chloride to a measurable extent under these conditions. However, bis(perfluoroaryl)- substituted 1d underwent a decrease in extinction coefficient in the presence of Bu4N+Cl− in THF (Fig. 3). The concentration dependence of the decrease in absorbance at 338 nm was fitted to a 1:1 binding isotherm, giving an association constant Ka of 310 ± 20 M−1 (Table 1). In a similar way, association constants were determined for chloride binding in acetone and acetonitrile: these were significantly lower than that determined in THF. Binding of Bu4N+Br− and Bu4N+BzO− by 1d was also observed in THF, but other anions resulted in decomposition (I−) or showed no evidence of binding (NO3−, toluenesulfonate (TsO−)).
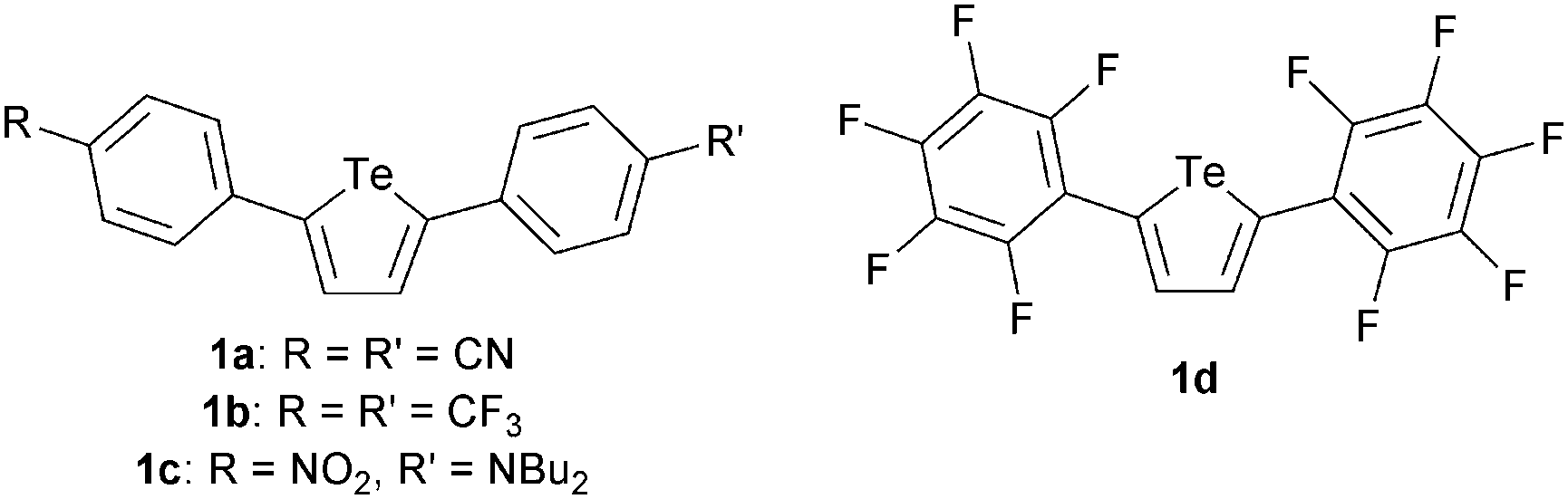 | ||
| Fig. 2 Structures of 2,5-diaryltellurophenes investigated for complexation of anions through chalcogen bonding. | ||
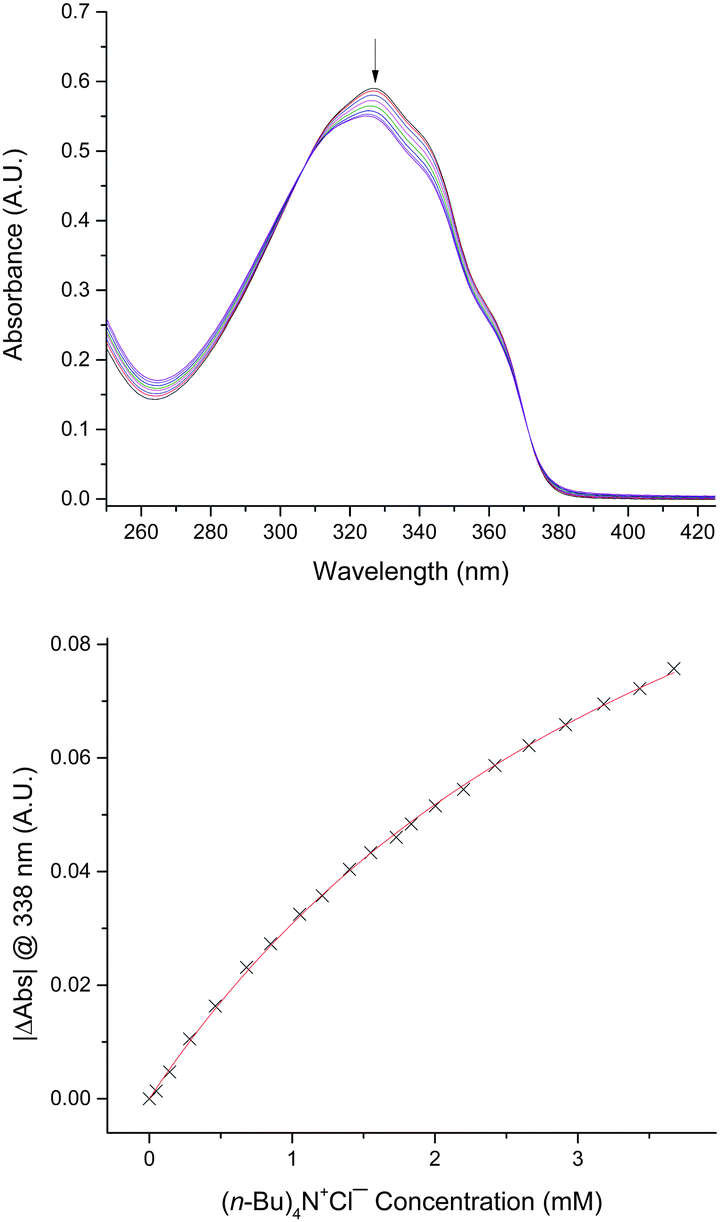 | ||
| Fig. 3 Changes in the optical absorbance spectrum of 1d upon addition of Bu4N+Cl− in THF at 298 K. | ||
| X− | Solvent | 1d | 2 |
|---|---|---|---|
a
K
a values were determined by fitting changes in optical absorbance as a function of concentration to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding isotherms. Each reported value is the average of at least three independent Ka determinations. The reported uncertainties reflect the standard deviation for each set of determinations.
b Not determined.
c Receptor underwent decomposition in the presence of Bu4N+X−. :1 binding isotherms. Each reported value is the average of at least three independent Ka determinations. The reported uncertainties reflect the standard deviation for each set of determinations.
b Not determined.
c Receptor underwent decomposition in the presence of Bu4N+X−.
|
|||
| Cl− | THF | 310 ± 20 | 2290 ± 30 |
| Cl− | Acetone | 6.2 ± 0.2 | 111 ± 1 |
| Cl− | MeCN | <3 | 19 ± 0.3 |
| Br− | THF | 103 ± 5 | 413 ± 5 |
| I− | THF | n.d.b,c | 107 ± 8c |
| BzO− | THF | 34 ± 3c | 66.4 ± 0.5c |
| NO3− | THF | <5 | 19.6 ± 0.9 |
| TsO− | THF | <2 | <2 |
The complex of 1d with Cl− was modelled using density functional theory (DFT). The calculations were carried out using Gaussian 09,17 with the dispersion-corrected B97-D3 functional,18 the Def2-TZVP basis set19 and the polarizable continuum model for THF solvent. The minimum-energy geometry is stabilized by a chalcogen bonding interaction having a Cl⋯Te distance of 3.07 Å and a 168° Cl⋯Te–C angle (Fig. 4). An anion–arene interaction20 with one of the perfluoroaryl groups appears to be present: the shortest distance between Cl− and the plane of the aromatic system is 3.35 Å, and the perfluoroarene group is oriented in a way that exposes the electron-deficient face to the anion (80° dihedral angle between perfluoraryl and tellurophene groups). The anion complexation mode is reminiscent of the calculated structure of a bromotelluronium bromide, one of the proposed intermediates in the oxidative addition of Br2 to 2,5-diphenyltellurophene.16 The calculated molecular electrostatic potential of 1d in the chloride-bound conformation shows regions of electron-deficiency at tellurium and at the centroid of the perfluoroaryl group (Fig. 4). It is likely that the higher π-acidity of the arene groups of 1d relative to 1a–1c contributes to its anion affinity: molecular electrostatic potential calculations indicate that the magnitudes of the σ-holes at Te are similar for 1a and 1d (see the ESI†), but only the latter showed evidence of Cl− binding. 2,5-Bis(pentafluorophenyl)thiophene,21 the analog of 1d having sulfur in place of tellurium, did not undergo any change in optical absorbance spectrum upon addition of Bu4N+Cl− in THF. The inability to determine an association constant for this control receptor prevented a quantitative assessment of the relative contributions of chalcogen bonding and anion–arene interactions to Cl− binding by the tellurophene congener. However, it indicates that chalcogen bonding contributes in an important way to the anion affinity of 1d.
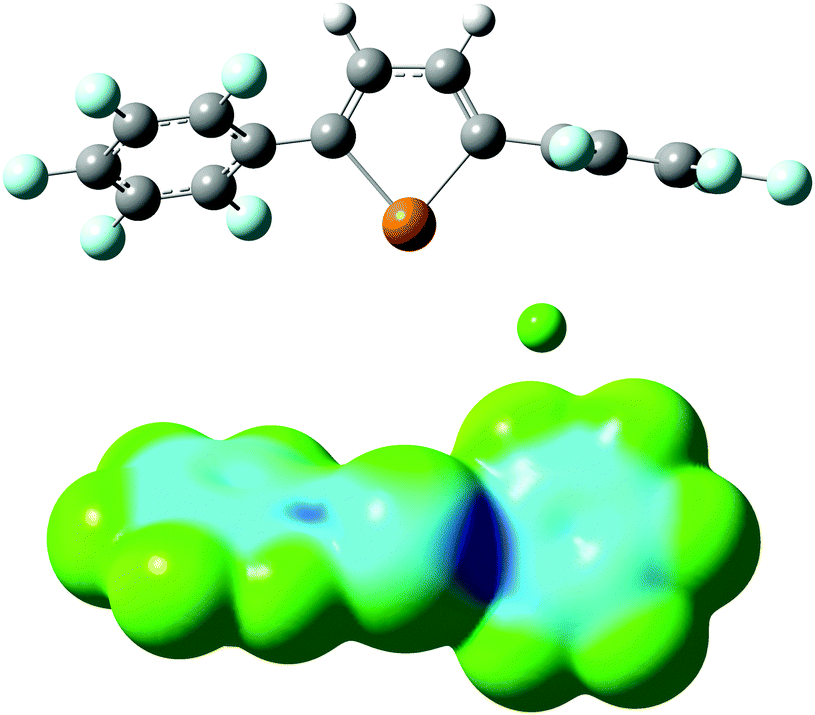 | ||
| Fig. 4 Calculated geometry (B97-D3/Def2-TZVP) of the 1d–Cl− complex; molecular electrostatic potential surface of 1d in its chloride-bound conformation. | ||
Having shown that 1d is capable of attractive interactions with anions, we sought to develop a bidentate anion receptor incorporating two tellurophene groups. Computational modelling suggested that an ethynylene spacer22,23 would place the two chalcogen bond donor groups at an appropriate distance for a two-point interaction with chloride. Receptor 2 was prepared from 2-iodo-5-(perfluorophenyl)tellurophene 4 by sequential Sonogashira couplings (Scheme 1).
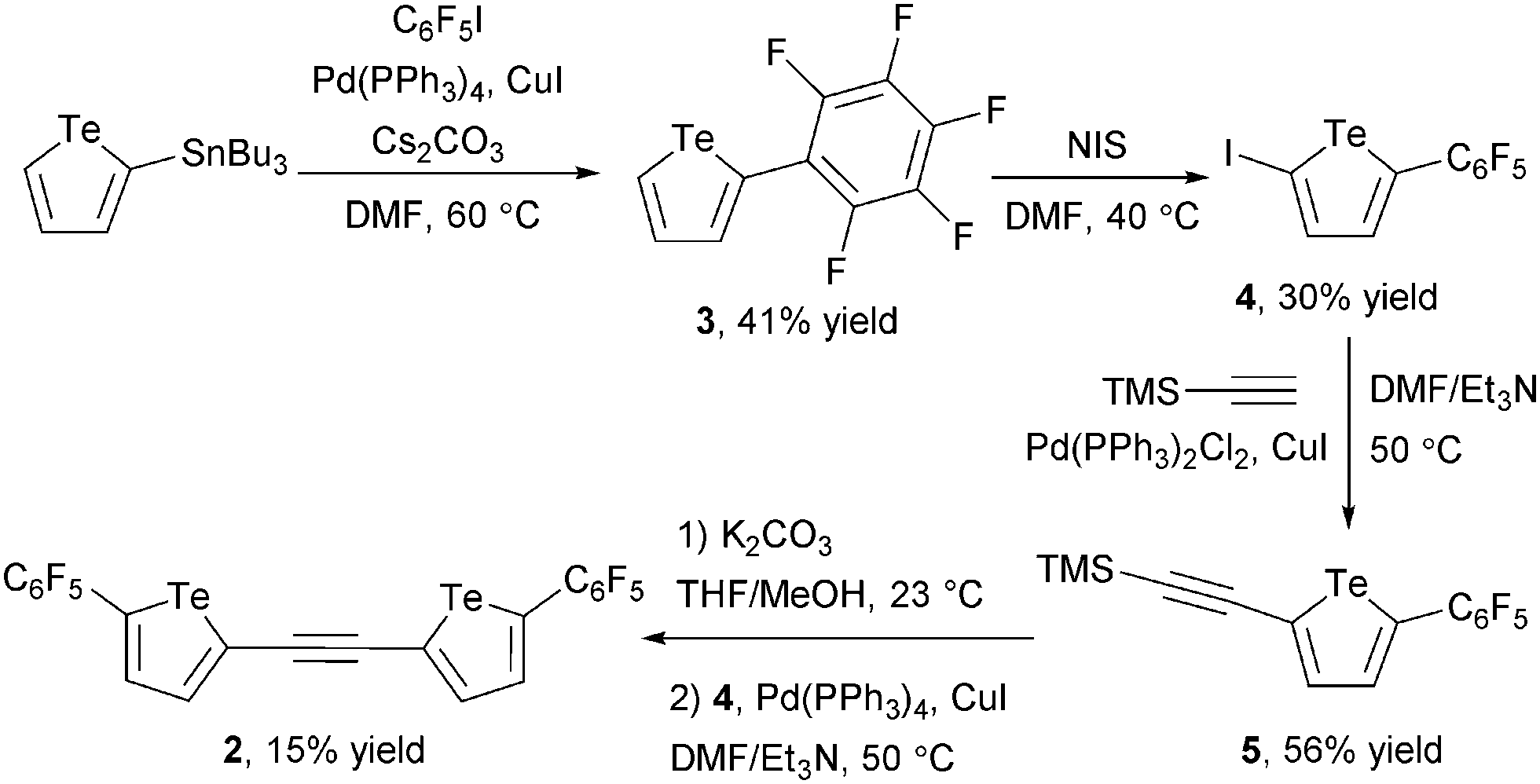 | ||
| Scheme 1 Synthesis of tellurophene-based bidentate anion receptor 2. | ||
Addition of Bu4N+Cl− to 2 in THF resulted in significant changes to the optical absorbance spectrum, with a decrease in intensity of the peaks at 371 and 403 nm and an increase in intensity at 422 nm (Fig. 5). Curve-fitting to a 1:1 model gave an association constant of 2290 M−1, roughly an order of magnitude higher than that determined for chloride binding by 1d. The 1:1 stoichiometry of Cl− complexation by 2 was supported by Job plot analysis of the absorbance data (see the ESI†). Nuclear magnetic resonance (NMR) spectroscopy in d8-THF provided further evidence for the interaction of 2 with Cl−: upfield changes in 1H NMR chemical shift and 19F NMR chemical shift were observed in the presence of Bu4N+Cl−. An association constant of 3200 ± 200 M−1 was determined from the 1H and 19F NMR titration data, in good agreement with the value determined by optical spectroscopy. Association constants of 2 with Cl− were also determined in acetone and acetonitrile. These were significantly lower than obtained in THF solvent, but enhancements in chloride affinity for 2versus1d were evident for each of the three solvents.
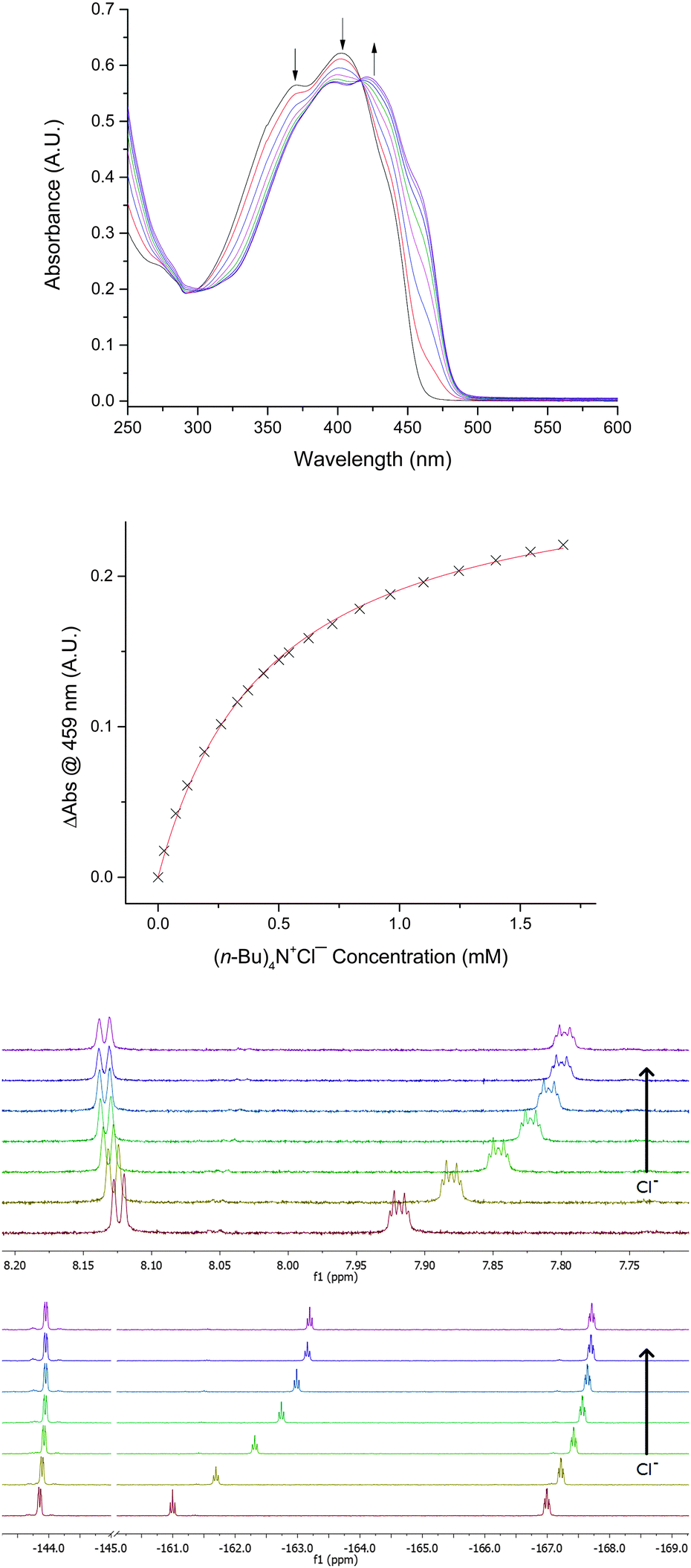 | ||
| Fig. 5 Changes in the optical absorbance spectrum, 1H NMR spectrum and 19F NMR spectrum of 2 upon addition of Bu4N+Cl− in THF at 298 K. | ||
The roughly tenfold increase in Ka for 2 relative to 1d is consistent with the hypothesis that the ethynylene-linked compound acts as a bidentate anion receptor. Indeed, the DFT-calculated minimum geometry for the 2–Cl− complex places the anion between the two tellurium atoms, with Cl⋯Te distances of 3.23 Å and Cl⋯Te–C angles of 170° (Fig. 6). This bidentate geometry does not allow for the type of anion–arene interaction that apparently stabilizes the 1d–Cl− complex. Thus, direct comparison with the 1d–Cl− association constant may underestimate the magnitude of the chelate effect in receptor 2, which appears to operate purely through chalcogen bonding. The value of Ka for Cl− binding by 2 in acetone (111 M−1) is roughly an order of magnitude lower that those for interactions of bidentate, iodoperfluoroarene-based halogen bond donors with Cl−, which are generally on the order of 103 M−1 in this solvent.23 Although the anion affinities of tellurophenes are relatively low in comparison to typically employed hydrogen or halogen bond donor groups, opportunities exist to enhance them through substitution, preorganization or incorporation of cationic substituents.
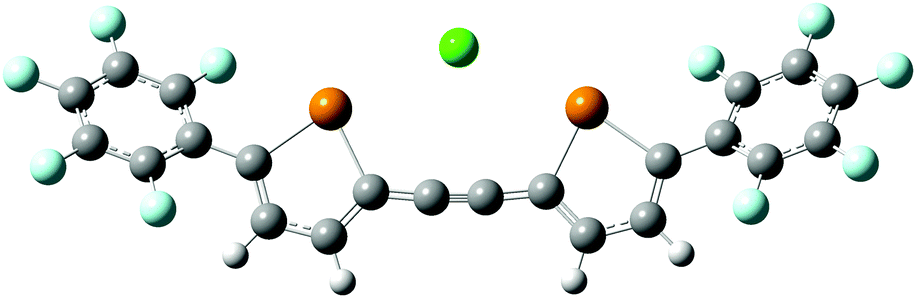 | ||
| Fig. 6 Calculated geometry (B97-D3/Def2-TZVP) of the 2–Cl− complex. | ||
Interactions of 2 with bromide, nitrate, benzoate and toluensulfonate were probed by optical spectroscopy (Table 1). The ethynylene-based bis(tellurophene) 2 has higher affinity for each of these anions than does compound 1d, although the magnitude of the chelate effect appears to be higher for chloride than for the other anions tested. The trend in association constants (Cl− > Br− > NO3−, TsO−) follows that observed for halogen bonding of iodoperfluoroarenes with anions in relatively nonpolar solvents (acetone, acetonitrile), and is in qualitative agreement with the calculated energies of complexes of 2 with these anions (see the ESI†).
In conclusion, we have shown that electron-deficient tellurophenes are able to bind to anions through chalcogen bonding interactions. The calculated modes of binding of tellurophene 1d and ethynylene-linked bis(tellurophene) 2 illustrate how substituents adjacent to the tellurium center can engage in additional attractive interactions with the anion. In the case of 1d, an appropriately oriented perfluoroaryl substituent participates in an anion–arene interaction. For receptor 2, the additional interaction is a second chalcogen bond, resulting in bidentate anion coordination through two Te⋯Cl− contacts. Bidentate chalcogen bonding in the 2–Cl− complex leads to an increase in association constant of an order of magnitude relative to 1d. The synthetic versatility of chalcogenophenes, along with their unique optoelectronic and materials properties,24 point towards further opportunities to exploit their chalcogen bond donor ability in molecular recognition and host–guest chemistry.
This work was supported by NSERC (Discovery Grant and Canada Research Chairs Programs, Graduate Scholarship to E. I. C.), the Canada Foundation for Innovation (projects #17545, #19119 and #24986), and the Ontario Ministry of Research and Innovation.
Notes and references
- M. Iwaoka and S. Tomoda, J. Am. Chem. Soc., 1996, 118, 8077–8084 CrossRef CAS; A. F. Cozzolino, I. Vargas-Baca, S. Mansour and A. H. Mahmoudkhani, J. Am. Chem. Soc., 2005, 127, 3184–3190 CrossRef PubMed; C. Bleiholder, D. B. Werz, H. Köppel and R. Gleiter, J. Am. Chem. Soc., 2006, 128, 2666–2674 CrossRef PubMed; M. E. Brezgunova, J. Lieffrig, E. Aubert, S. Dahaoui, P. Fertey, S. Lebègue, J. G. Ángyán, M. Fourmigué and E. Espinosa, Cryst. Growth Des., 2013, 13, 3283–3289 Search PubMed.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and T. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- R. E. Rosenfeld Jr., R. Parthasarathy and J. D. Dunitz, J. Am. Chem. Soc., 1977, 99, 4860–4862 CrossRef CAS; T. N. Guru Row and R. Parthasarathy, J. Am. Chem. Soc., 1981, 103, 477–479 CrossRef; F. T. Burling and B. M. Goldstein, J. Am. Chem. Soc., 1992, 114, 2313–2320 CrossRef.
- A. F. Cozzolino, P. J. W. Elder and I. Vargas-Baca, Coord. Chem. Rev., 2011, 255, 1426–1438 CrossRef CAS.
- A. F. Cozzolino, J. F. Britten and I. Vargas-Baca, Cryst. Growth Des., 2006, 6, 181–186 CAS.
- A. F. Cozzolino, P. S. Whitfield and I. Vargas-Baca, J. Am. Chem. Soc., 2010, 132, 17265–17270 CrossRef CAS PubMed.
- A. Kremer, A. Fermi, N. Biot, J. Wouters and D. Bonifazi, Chem. – Eur. J., 2016, 22, 5665–5675 CrossRef CAS PubMed.
- P. C. Ho, P. Szydlowski, J. Sinclair, P. J. W. Elder, J. Kübel, C. Gendy, L. M. Lee, H. Jenkins, J. F. Britten, D. R. Morim and I. Vargas-Baca, Nat. Commun., 2016, 7, 11299 CrossRef CAS PubMed.
- T. M. Beale, M. G. Chudzinski, M. G. Sarwar and M. S. Taylor, Chem. Soc. Rev., 2013, 42, 1667–1680 RSC; L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7188–7195 CrossRef CAS PubMed.
- H. Zhao and F. Gabbaï, Nat. Chem., 2010, 2, 984–990 CrossRef CAS PubMed.
- N. A. Semenov, A. V. Lonchakov, N. A. Kushkarevsky, E. A. Suturina, V. V. Korolev, E. Lork, V. G. Vasiliev, S. N. Konchenko, J. Beckmann, N. P. Gritsan and A. V. Zibarev, Organometallics, 2014, 33, 4302–4314 CrossRef CAS.
- G. E. Garrett, G. L. Gibson, R. N. Straus, D. S. Seferos and M. S. Taylor, J. Am. Chem. Soc., 2015, 137, 4126–4133 CrossRef CAS PubMed.
- J. S. Murray, P. Lane, T. Clark and P. Politzer, J. Mol. Model., 2007, 13, 1033–1038 CrossRef CAS PubMed.
- W. Mack, Angew. Chem., Int. Ed., 1966, 10, 896 CrossRef.
- Y. S. Park, T. S. Kale, C.-Y. Nam, D. Choi and R. B. Grubbs, Chem. Commun., 2014, 50, 7964–7967 RSC.
- E. I. Carrera, A. E. Lanterna, A. J. Lough, J. C. Scaiano and D. S. Seferos, J. Am. Chem. Soc., 2016, 138, 2678–2689 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 09, Rev. D.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- B. L. Schottel, H. T. Chifotides and K. R. Dunbar, Chem. Soc. Rev., 2008, 37, 68–83 RSC; B. P. Hay and V. S. Bryantsev, Chem. Commun., 2008, 2417–2428 RSC; L. M. Salonen, M. Ellermann and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 4808–4842 CrossRef CAS PubMed; A. Frontera, P. Gamez, M. Mascal, T. J. Mooibroek and J. Reedijk, Angew. Chem., Int. Ed., 2011, 50, 9564–9583 CrossRef PubMed.
- K. Takimiya, N. Niihara and T. Otsubo, Synthesis, 2005, 10, 1589–1592 CrossRef.
- Y. Ducharme and J. D. Wuest, J. Org. Chem., 1988, 53, 5789–5791 CrossRef.
- M. G. Sarwar, B. Dragisic, E. Dimitrijevic and M. S. Taylor, Chem. – Eur. J., 2013, 19, 2050–2058 CrossRef CAS PubMed.
- J. Hollinger, D. Gao and D. S. Seferos, Isr. J. Chem., 2014, 54, 440–453 CrossRef CAS; E. I. Carrera and D. S. Seferos, Dalton Trans., 2015, 44, 2092–2096 RSC; E. Rivard, Chem. Lett., 2015, 44, 730–736 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Protocols for the synthesis of 2, NMR spectral data for 2, anion titration data and analysis, and computational details. See DOI: 10.1039/c6cc04818h |
| This journal is © The Royal Society of Chemistry 2016 |