
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Trehalose-based Janus cyclooligosaccharides: the “Click” synthesis and DNA-directed assembly into pH-sensitive transfectious nanoparticles†
J. L.
Jiménez Blanco
*a,
F.
Ortega-Caballero
a,
L.
Blanco-Fernández
b,
T.
Carmona
c,
G.
Marcelo
c,
M.
Martínez-Negro
d,
E.
Aicart
d,
E.
Junquera
*d,
F.
Mendicuti
*c,
C.
Tros de Ilarduya
*b,
C.
Ortiz Mellet
 a and
J. M.
García Fernández
*e
a and
J. M.
García Fernández
*e
aDepartment of Organic Chemistry, University of Sevilla, E-41012 Sevilla, Spain
bDepartment of Pharmacy and Pharmaceutical Technology, University of Navarra, E-31008 Pamplona, Spain
cDepartment of Analytical Chemistry, Physical Chemistry and Chemical Engineering, Universidad de Alcalá, E-28871 Alcalá de Henares, Spain
dDepartment of Physical Chemistry I, Universidad Complutense, E-28040 Madrid, Spain
eInstituto de Investigaciones Químicas (IIQ), CSIC – Universidad de Sevilla, E-41092 Sevilla, Spain. E-mail: jogarcia@iiq.csic.es
First published on 30th June 2016
Abstract
The convergent preparation of Janus molecular nanoparticles by thiourea-“clicking” of α,α′-trehalose halves has been implemented; the strategy allows access to macrocyclic derivatives with seggregated cationic and lipophilic domains that in the presence of DNA undergo pH-dependent self-assembly into lamellar superstructures, as established by electrochemical, structural (SAXS), microscopical (TEM) and computational techniques, that mediate transfection in vitro and in vivo.
The term “Janus” was initially adopted from Roman mythology by materials scientists to denote particles with a three-dimensionally dissymmetrical distribution of physical/chemical features.1 Because of their morphological and chemical asymmetry, Janus particles can assemble into unique superstructures that cannot be obtained via the assembly of homogeneous particles.2 The notion has been further extended to describe the entities exhibiting dichotomy in general, from covalent structures to supramolecular assemblies and nanocomposites. Among them, molecular Janus architectures are particularly interesting. Their decreased sizes of typically only a few nanometers and the possibility of precise chemical tailoring make them ideally suited for the incorporation of functional and structural hierarchy. However, most of the molecular Janus entities reported so far are based on dendritic3 or polymeric scaffolds4 with a high degree of conformational flexibility. Alternatively, connecting together hydrophobic and hydrophilic 3D molecular frameworks with precisely defined chemical structures, sizes, shapes, symmetry and surface functional groups, referred to as “molecular nanoparticles” (MNPs), have been used to generate new monodisperse Janus-like topologies.5 The examples on record include snowman or dumbbell-like nanostructures obtained from polyhedral oligomeric silsesquioxane (POSS), C60 fullerene, polyoxometalate (POM), cyclodextrin (CD) or calixarene (CA) building blocks that still keep a relatively high degree of conformational flexibility and in which the spatial separation between the components fluctuates depending on the nature of the tether.6 “Monomeric” Janus-like MNPs, better mimicking “hard” Janus objects, can be accessed by selective facial functionalization when the starting precursor exhibit intrinsic molecular anisotropy, as is the case for CD and CA platforms.7 Recently, Chiara and coworkers8 have reported an elegant strategy to break the symmetry and create a “perfect” Janus feature in a cubic POSS precursor by exploiting the geometrical complementarity of the cube face with a conveniently tetra-functionalized partner. In any case, with a few exceptions such divergent approaches require a multiple-step synthesis involving delicate control over reaction conditions and nontrivial purifications to warrant diasterochemical purity. Here we present an alternative convergent tactic based synthesis in the multipoint covalent assembly of dissimilar halves that allows elaborating “perfect” Janus cyclooligosaccharides made of α,α′-trehalose (cyclotrehalans, CTs) whose self-assembly behaviors are dictated by symmetry breaking in both functional groups and supramolecular interactions. The methodology has been applied to the efficient synthesis of MNPs which is capable of reversibly condensing DNA into self-assembled lamellar nanocomplexes (CTplexes) with transfection-mediating capabilities in vitro and in vivo (Fig. 1).
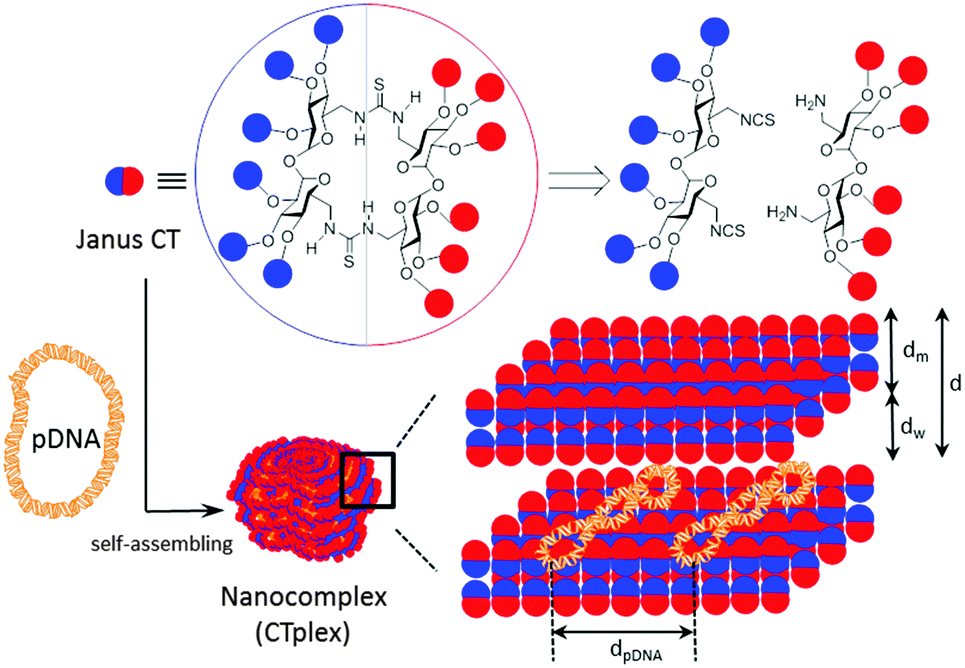 | ||
| Fig. 1 Schematic representation of Janus cyclooligosaccharides obtained using thiourea-“clicking” of α,α′-trehalose halves. In the cartoon, red circles represent cationic groups and blue circles lipophilic substituents. Hierarchical assembly in the presence of plasmid DNA to give nanocomplexes (CTplexes) with a lamellar structure is represented. | ||
In contrast to other macrocyclic MNPs such as CDs or CAs, canonic CTs exhibit identical faces that are brought together after a very efficient macrocyclization step involving a double “click”-type thiourea-forming reaction.9 The concave shape of the constitutive α,α′-trehalose moieties, dictated by the concurrence of two exo-anomeric effects at the glycosidic linkages, preorganizes the disaccharide to favour macrocyclic over oligomeric structures upon bridging through the primary positions. Thiourea tethering further reinforces macrocyclization efficiency: after the first isothiocyanate-amine coupling, the formation of a seven-membered intramolecular hydrogen bond stabilizes the Z,E-rotamer and places the next reacting groups in close proximity to zip the macroring. We envisioned that stacking of amphiphilic molecular Janus CTs composed of hydrophobic and cationic moieties would be prohibited by the repulsive interactions introduced by the charges on the surface. In the presence of an oligonucleotide chain, the interplay of attractive coulombic and hydrophobic interactions may promote DNA-directed assembly into nanocomplexes whose stability would depend on the protonation extent, which may be used for pH-sensitive non-viral gene delivery.
To check the above working hypothesis, the diisothiocyanate and diamine precursors 1 and 2, bearing respectively six hexanoyl tails and six protected cationizable amine groups at the secondary hydroxyls, were synthesized (ESI†) and reacted (→3) to give, after deprotection, the multihead-multitail Janus CT 4 in over 80% yield. Further elaboration of this prototype was performed by thiourea coupling of 4 with the branching building block 5(→6), affording the dendritic adduct 7 (Scheme 1). Both compounds were purposely designed to incorporate structural elements (hexanoyl tails, thiourea H-bond donor centres, multivalent/dendritic amine clusters) previously found advantageous in the design of molecular artificial viruses.10
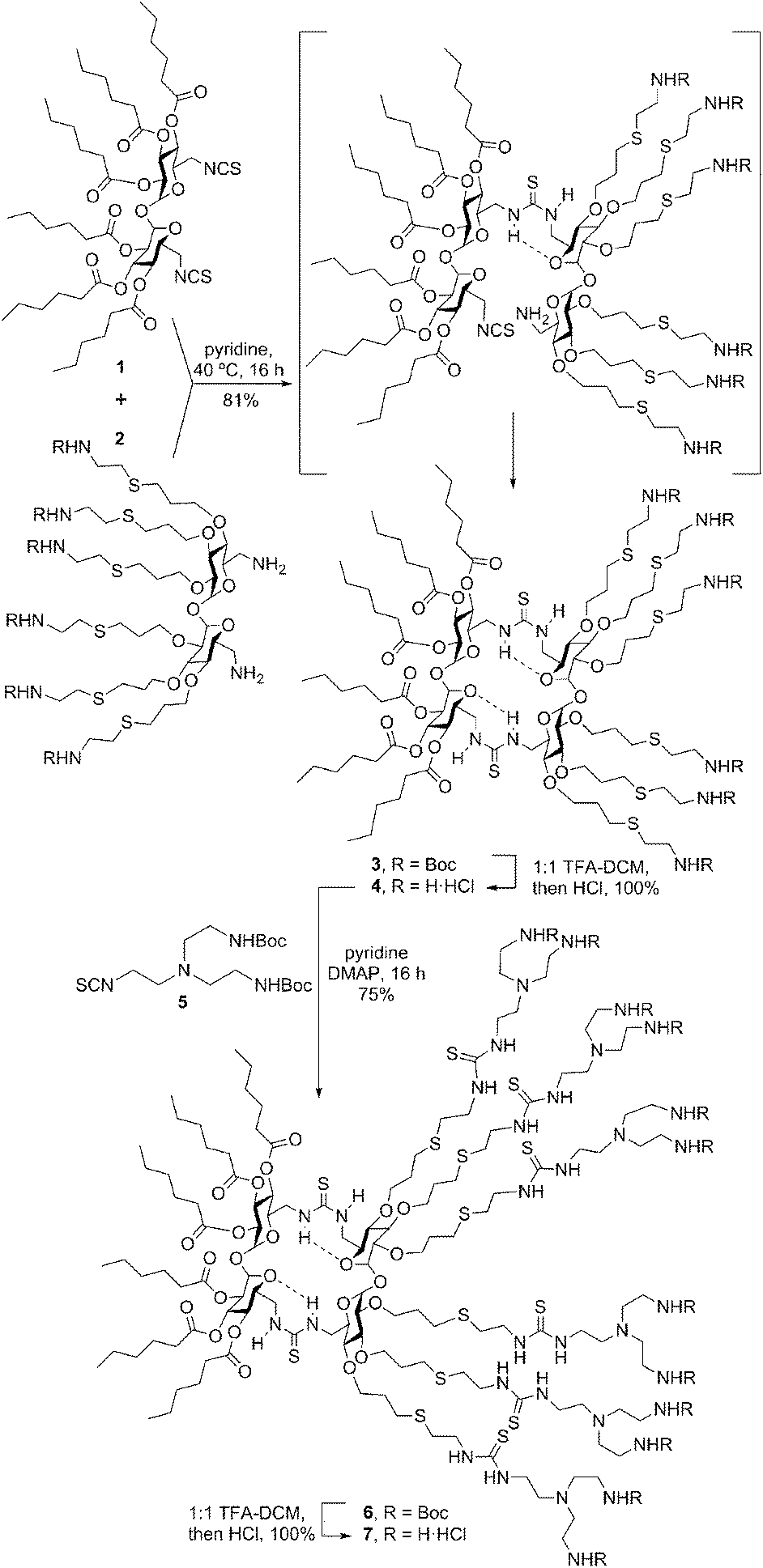 | ||
| Scheme 1 Synthesis of the Janus cyclotrehalans 4 and 7. | ||
The effective charges available for coulombic interaction between the Janus CTs and DNA, determined from ζ-potential measurements, were found to be significantly different from those expected considering the ionizable N and P atoms in each partner. For instance, CT 4 exhibited positive net charges that are around 75% of the nominal ones in the presence of DNA (Table S1, ESI†). Contrary to linear double helix DNA, which keeps its negative charge (−2 per base pair) totally available for the gene vector,11 plasmid DNA rendered an available negative charge per bp of around 7% of its nominal value when being compacted by CT 4. It means that the effective charge ratios, ρeff, are around 10- to 11-fold the nominal charge ratios ρnom (also named as N/P). These data support the fact that the plasmid remains in a supercoiled conformation under physiological conditions, retaining an important percentage of its cationic sodium counter ions after interaction with the vector.
The circular dichroism spectra of DNA registered in the absence and in the presence of compounds 4 or 7 at pH 7.4 unequivocally evidenced the existence of interactions that distorted the typical B-type structure of uncomplexed DNA, probably to a Z-DNA form. Thus, a concentration-dependent decrease in the intensities of the positive and negative bands at 278 and 245 nm, arising from stacking interactions between bases and from polynucleotide helicity, respectively, was initially observed (Fig. 2A and Fig. S14, ESI†), with a change in sign at CT:DNA mass molar ratios higher than 2 (Fig. 2B).
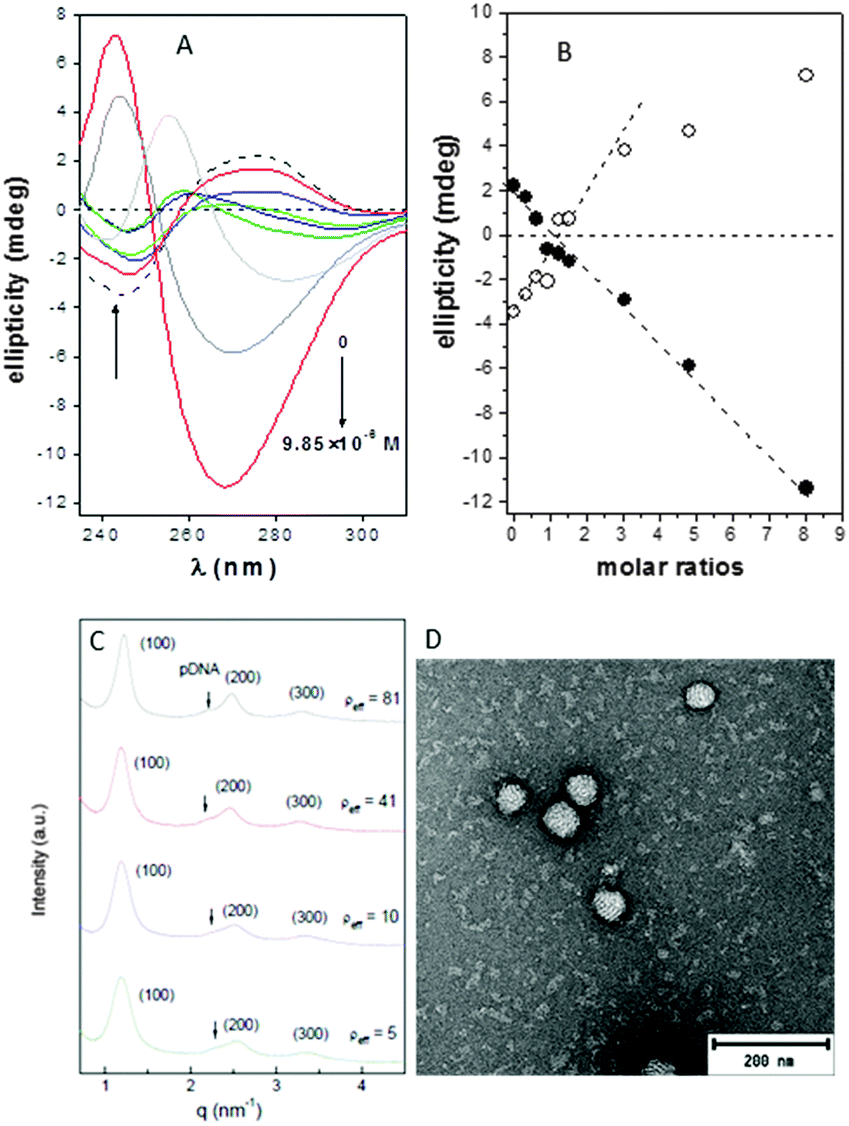 | ||
| Fig. 2 (A) Circular dichroism spectra in the 230–325 nm region for solutions containing Janus CT 4 (0, 0.37, 0.75, 1.12, 1.50, 1.87, 3.74, 5.91 and 9.85 μM) and DNA fixed 1.23 μM concentration in HEPES at 25 °C. (B) Ellipticity values as a function of the 4:DNA mass molar ratios for the maxima of the two bands appearing near 245 nm (open symbols) and 275 nm (filled symbols). (C) SAXS diffractograms for 4:pDNA complexes formulated at several effective charge ratios (ρeff). (D) TEM micrograph of 4:pDNA complexes formulated at N/P 10. | ||
Small-angle X-ray scattering (SAXS) diffractograms (intensity vs. q factor) of 4:pDNA complexes at several effective charge ratios (ρeff = 5, 10, 41 and 81, i.e. N/P ratios 0.45, 0.91, 3.7 and 7.4, respectively) showed three peaks that were well indexed to a lamellar lyotropic liquid crystal phase, Lα, regardless of ρeff (Fig. 2C). This lamellar arrangement, known to be correlated with potentially high transfection efficiencies,12 arises from the self-assembly of the Janus CT molecules into lipidic bilayers in the confined space between quasi-parallel DNA supercoils, with thicknesses represented by dm, and dw, respectively, being d = dm + dw. The characteristic interlayer distances, d, directly related to the q factor (d = 2πn/qhkl, n is the diffraction order) remains constant with ρeff at 5.2 ± 0.1 nm (Fig. 2C and Fig. S13, ESI†). Considering these d values and the fact that pDNA supercoils need around dw ∼ 2.5 nm to be sandwiched by CT bilayers, it can be deduced that the thickness of the bilayer, dm, must be around 2.7 nm. TEM micrographs (Fig. 2D) of the CTplexes also showed the aggregates with a clear multilamellar pattern, in full agreement with SAXS results.
To get a deeper insight into the interactions that govern the hierarchical process that leads to CTplex formation, the stability of a head-to-head dimer of 4, the smallest unit of the bilayer arrangement, was first studied by molecular mechanics (MM) in explicit water. The minimum binding energy (MBE) structure was initially obtained by sequentially approaching two Janus CT molecules in the corresponding perhydrochloride form, with their center of mass on the y axis in a coordinate system (Fig. S16, ESI†). The binding energy profile corresponded to a structure in which the lipidic hexanoyl chains significantly intercrossed and the cationic arms onto the opposite face of the Janus macrocycle adopted an open bouquet disposition to avoid electrostatic repulsions and steric clashes (Fig. S17, ESI†). The MBE structure was then placed between two symmetrically located and oriented B DNA helix fragments containing twelve nucleotides (CGCGAATTCGCG) and the oligonucleotide chains were approached simultaneously in steps of 1 Å (Fig. 3) through the major groove. The binding energies obtained using this procedure, which were rather favorable, led to an efficient packing. Remarkably, the distance between the centers of mass of the nitrogen clusters in the CTs, which would represent a measurement of the CT bilayer thickness, was reduced from near ∼3.3 nm for the isolated CT dimer to ∼2.6 nm for the most stable structure of the CTplex, very well fitting the above SAXS experimental data. The most stable CTplex structure generated (Fig. 3) was used as the starting conformation for 1.0 ns MD simulations, which confirmed the stability of the nanocomplex through the trajectory (Fig. S20 and S21, ESI†). Interestingly, when calculations were conducted on a fully cationized form of 4 (by removing the chloride anions) the dimer became unstable. We speculated that increasing protonation in the acidic environment of the endosomes after CTplex cellular uptake would then destabilize the Janus CT bilayers, contributing both to endosome disruption and intracellular DNA release, thereby facilitating the transfection process.13
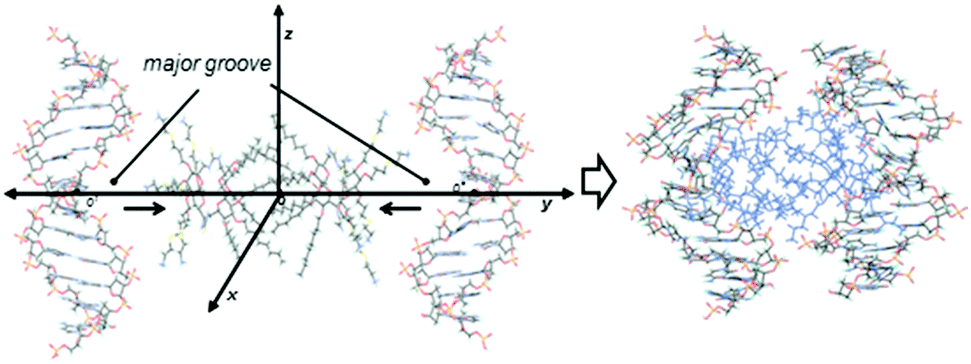 | ||
| Fig. 3 Coordinate system used for DNA fragments approaching the MBE structure of the CT 4 dimer along the y coordinate by the major groove (left) and the MBE structure of the resulting CT:DNA complex (right). | ||
Electrophoretic mobility experiments in the agarose gel followed by visualization using the fluorescent intercalating agent GelRed® confirmed that full pDNA complexation was achieved with both Janus CTs 4 and 7 at N/P ratios >1 (ρeff > 10). After treatment with a nuclease and dissociation with sodium dodecylsulfate (SDS), intact DNA could be recovered from CTplexes, demonstrating that the DNA cargo was protected from the environment following self-assembly (Fig. S22, ESI†). CTplexes formulated with 4 and 7 at N/P ratios 5 and 10, (hydrodynamic diameter 123 to 230 nm; ζ-potential +18 to +30 mV. See the ESI,† Table S3), promoted transfection in African green-monkey epithelial kidney COS-7 cells (Fig. 4A) and human hepatocellular carcinoma HepG2 cells (Fig. S22, ESI†) with efficiencies that were comparable to those obtained with polyplexes formulated with branched poly(ethyleneimine) (bPEI), a commonly used positive control for nonviral gene delivery, even in the presence of serum. It is noteworthy that no toxicity was observed for any CTplex formulation, compared with 60–70% cell viability for bPEI polyplexes. The nanocomplexes formulated with 4 and 7 at N/P 10 were next injected systemically into mice, and their activity was compared with control PBS and the naked DNA. The results, based on the luciferase reporter gene expression, indicated that 24 h after the intravenous administration of the CTplexes transfection occurs mainly in the liver and lung (Fig. 4B), with negligible luminescence detected in other organs. Increases at transfection levels relative to the naked DNA were above one and two orders of magnitude for 4:pDNA and 7:pDNA CTplexes, respectively. The advantages of the convergent synthetic methodology in terms of versatility and ease of manipulation of the precursors should now allow the preparation of a larger collection of monodisperse Janus MNPs for structure/self-assembly/biological activity relationship studies in view of developing optimized nanodevices programmed for biomedical applications.
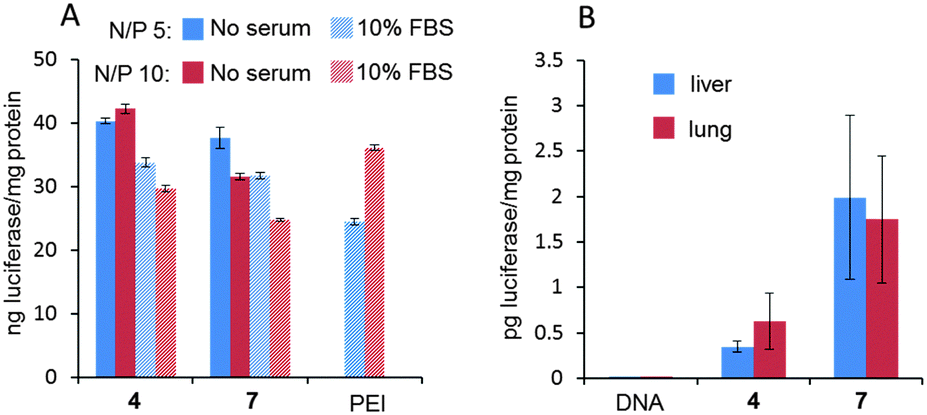 | ||
| Fig. 4 (A) Transfection efficiency in COS-7 cells for CTplexes formulated with Janus CTs 4 or 7 and the luciferase-encoding reporter gene pCpG-hCMV-SPEC-eFLuc at N/P ratios 5 and 10 in the absence and presence of 10% fetal bovines serum (FBS). Data obtained with bPEI polyplexes (N/P = 5 and 10, 10% FBS) under identical conditions are included for comparison. The data represent the mean ± SD of three wells and are representative of three independent determinations. (B) Gene expression conducted in vivo after intravenous administration of 60 μg of pCpG-hCMV-SPEC-eFLuc formulated as CTplexes with 4 and 7 at a N/P 10. Bars represent the mean ± SD (n = 7 animals). | ||
The authors thank MINECO (contract numbers CTQ2012-30821, SAF2013-44021-R, CTQ2015-64425-C2-1-R and CTQ2015-64425-C2-2-R), the Junta de Andalucía (contract number FQM2012-1467), University Complutense of Madrid (project no. UCMA05-33-010), the Government of Navarra (Department of Innovation and Industry, contract number IIQ14334.RI1), the University of Navarra Foundation (FUN), and the European Regional Development Funds (FEDER and FSE) for financial support. SAXS experiments were performed at NCD11 beamline at ALBA Synchrotron Light Facility with the collaboration of ALBA staff. The CITIUS (Univ. Sevilla) is also thanked for technical support.
Notes and references
- (a) P. G. de Gennes, Science, 1992, 256, 495 CAS; (b) A. Walther and A. H. E. Müller, Chem. Rev., 2013, 113, 5194 CrossRef CAS PubMed; (c) Y. Song and S. Chen, Chem. – Asian J., 2014, 9, 418 CrossRef CAS PubMed.
- Y. Liu, B. Liu and Z. Nie, Nano Today, 2015, 10, 278 CrossRef CAS.
- (a) V. Percec, D. A. Wilson, P. Leowanawat, C. J. Wilson, A. D. Hughes, M. S. Kaucher, D. A. Hammer, D. H. Levine, A. J. Kim, F. S. Bates, K. P. Davis, T. P. Lodge, M. L. Klein, R. H. DeVane, E. Aqad, B. M. Rosen, A. O. Argintaru, M. J. Sienkowska, K. Rissanen, S. Nummelin and J. Ropponen, Science, 2010, 328, 1009 CrossRef CAS PubMed; (b) S. Zhang, H.-J. Sun, A. D. Hughes, B. Draghici, J. Lejnieks, P. Leowanawat, A. Bertin, L. O. De Leon, O. V. Kulikov, Y. Chen, D. J. Pochan, P. A. Heiney and V. Percec, ACS Nano, 2014, 8, 1554 CrossRef CAS PubMed; (c) H.-J. Sun, S. Zhang and V. Percec, Chem. Soc. Rev., 2015, 44, 3900 RSC.
- (a) M. Zhang and A. H. E. Müller, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 3461 CrossRef CAS; (b) L. Cheng, G. Hou, J. Miao, D. Chen, M. Jiang and L. Zhu, Macromolecules, 2008, 41, 8159 CrossRef CAS; (c) B. Nandan and A. Horechyy, ACS Appl. Mater. Interfaces, 2015, 7, 12539 CrossRef CAS PubMed; (d) J. Liu and J. Huskens, Chem. Commun., 2015, 51, 2694 RSC.
- W.-B. Zhang, X. Yu, C.-L. Wang, H.-J. Sun, I.-F. Hsieh, Y. Li, X.-H. Dong, K. Yue, R. Van Horn and S. Z. D. Cheng, Biomacromolecules, 2014, 47, 1221 CrossRef CAS.
- (a) Y. Li, W.-B. Zhang, I.-F. Hsieh, G. Zhang, Y. Cao, X. Li, C. Wesdemiotis, B. Lotz, H. Xiong and S. Z. D. Cheng, J. Am. Chem. Soc., 2011, 133, 10712 CrossRef CAS PubMed; (b) H. Liu, C.-H. Hsu, Z. Lin, W. Shan, J. Wang, J. Jiang, M. Huang, B. Lotz, X. Yu, W.-B. Zhang, K. Yue and S. Z. D. Cheng, J. Am. Chem. Soc., 2014, 136, 10691 CrossRef CAS PubMed; (c) L. Gallego-Yerga, M. Lomazzi, F. Sansone, C. Ortiz Mellet, A. Casnati and J. M. García Fernández, Chem. Commun., 2014, 50, 7440 RSC.
- (a) V. Bagnacani, V. Franceschi, M. Bassi, M. Lomazzi, G. Donofrio, F. Sansone, A. Casnati and R. Ungaro, Nat. Commun., 2013, 4, 1721 CrossRef PubMed; (b) Y. Li, Y. Qian, T. Liu, G. Zhang, J. Hu and S. Liu, Polym. Chem., 2014, 5, 1743 RSC; (c) L. Gallego-Yerga, M. Lomazzi, V. Franceschi, F. Sansone, C. Ortiz Mellet, G. Donofrio, A. Casnati and J. M. García Fernández, Org. Biomol. Chem., 2015, 13, 1708 RSC.
- A. Blázquez-Moraleja, M. E. Pérez-Ojeda, J. R. Suárez, M. L. Jimeno and J. L. Chiara, Chem. Commun., 2016, 52, 5792 RSC.
- (a) J. M. Benito, J. L. Jiménez Blanco, C. Ortiz Mellet and J. M. García Fernández, Angew. Chem., Int. Ed., 2002, 41, 3674 CrossRef CAS; (b) D. Rodríguez-Lucena, J. M. Benito, E. Álvarez, C. Jaime, J. Perez-Miron, C. Ortiz Mellet and J. M. García Fernández, J. Org. Chem., 2008, 73, 2967 CrossRef PubMed; (c) D. Rodríguez-Lucena, C. Ortiz Mellet, C. Jaime, K. K. Burusco, J. M. García Fernández and J. M. Benito, J. Org. Chem., 2009, 74, 2997 CrossRef PubMed.
- C. Ortiz Mellet, J. M. Benito and J. M. García Fernández, Chem. – Eur. J., 2010, 16, 6728 CrossRef CAS PubMed.
- E. Junquera and E. Aicart, Curr. Top. Med. Chem., 2014, 14, 649 CrossRef CAS PubMed.
- M. Muñoz-Ubeda, S. K. Misra, A. L. Barran-Berdon, C. Aicart-Ramos, M. B. Sierra, J. Biswas, P. Kondaiah, E. Junquera, S. Bhattacharya and E. Aicart, J. Am. Chem. Soc., 2011, 133, 18014 CrossRef PubMed.
- L. Gallego-Yerga, L. Blanco-Fernández, K. Urbiola, T. Carmona, G. Marcelo, J. M. Benito, F. Mendicuti, C. Tros de Ilarduya, C. Ortiz Mellet and J. M. García Fernández, Chem. – Eur. J., 2015, 21, 12093 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details (synthesis, electrochemical determinations, nanoparticle characterisation, computational studies, in vitro and in vivo transfection) and copies of the NMR spectra of the new compounds. See DOI: 10.1039/c6cc04791b |
| This journal is © The Royal Society of Chemistry 2016 |