
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The Pauson–Khand reaction using alkynylboronic esters: solving a long-standing regioselectivity issue†
Thierry
León
*a and
Elena
Fernández
*b
aCenter for Chemical Technology of Catalonia (CTQC), Marcel·lí Domingo, s/n, Edifici N5, 43007 Tarragona, Spain. E-mail: thierry.leon@fundacio.urv.cat
bDepartament de Química Física i Inorgànica, Universitat Rovira i Virgili, Marcel·lí Domingo, s/n, Edifici N4, 43007 Tarragona, Spain. E-mail: mariaelena.fernandez@urv.cat
First published on 24th June 2016
Abstract
The first intermolecular Pauson–Khand reaction, conducted using internal alkynylboronic esters, allows the installation of the boronic ester moiety at the β-position of the cyclopentenone with total regio- and stereoselectivity.
Since its first report by Khand and Pauson in the early seventies,1 the Pauson–Khand reaction (PKR) has proven to be a very powerful tool to build cyclopentenone cores. The reaction consists of a [2+2+1] cycloaddition involving an alkyne, an alkene and carbon monoxide allowing the assembly of architecturally complex molecules from very simple starting materials.2 The intermolecular version has to face challenging issues namely the regioselective addition of alkenes and alkynes. Towards this end, terminal alkynes have been successfully employed yielding the α-substituted adduct in a regioselective manner (Scheme 1, route a). However, internal nonsymmetric alkynes can potentially provide both regioisomers.3 Interestingly, when sterically or electronically biased internal alkynes are used, the largest or the most electrondonating group (EDG) is mainly (but not exclusively) placed at the α-position of the resulting adduct (Scheme 1, route b). However, when non-biased alkynes are used, mixtures of α/β-substituted adducts are observed (Scheme 1, route c).4 This fact has been supported by recent computational studies where the lack of electronic or steric differentiation along the triple bond does not allow, in some cases, a clear distinction in the regioselective outcome of the reaction.5 Bearing these limitations in mind, we envisioned the possibility of using boron chemistry to solve the lack of selectivity in the PKR reaction for internal alkynes, complementing the current approaches to prepare α,β-diarylcyclopentenones, namely the Nazarov reaction,6 Ni- and Pd-catalysed annulations7 or Au-catalyzed [4+1] cyclizations.8
 | ||
| Scheme 1 Regioselective scenarios of alkynes in intermolecular PKR. | ||
Our proposal is based on a two-step strategy and its usefulness lies in the use of internal alkynylboronic esters, which take advantage of the polarization along the triple bond falling into a specific case of biased internal alkynes (Scheme 1, route d). Internal alkynylboronic esters are versatile synthons that have been successfully used in a vast array of reactions such as standard Suzuki–Miyaura cross-coupling reactions9 or cycloaddition reactions,10 but its use in the PKR is surprisingly unprecedented in the literature.
Herein, we describe, the first PKR using internal alkynylboronic esters, with the exclusive formation of a single exo stereoisomer and the β-regioisomer. Subsequently, the B-substituted cyclopentenones are subjected to the Suzuki–Miyaura cross-coupling (SMC) allowing the overall sequential installation of α and β substitutions. This strategy allows access to both regioisomers by simply reversing the order of introduction of the substituents, converting it to a very direct and intuitive tool (Scheme 2).
 | ||
| Scheme 2 Selection of the regioisomer by a sequential strategy. | ||
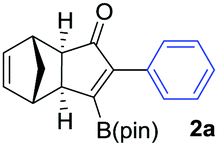
We initiated our study by optimizing the PKR using 4,4,5,5-tetramethyl-2-(phenylethynyl)-1,3,2-dioxaborolane (1a) as a model substrate (Table 1). We found that the optimal reaction conditions were the combination of Co2(CO)8 (1 equiv.), norbornadiene (NBD, 3 equiv.), and 4-methylmorpholine N-oxide (NMO, 6 equiv.) in dichloromethane (0.03 M) at room temperature yielding the corresponding adduct 2a in 75% isolated yield (Table 1, entry 1). Along with the desired product 2a, the protodeborylated side-product 3 was obtained in 9% yield. However, when thermal conditions were used in toluene, the yield of adduct 2a dropped to 38% together with 12% yield of side-product 3 (Table 1, entry 2), probably due to decomposition of intermediate species during the reaction. Attempts to stabilize the cobalt complex with PPh3 had a detrimental impact on the yield lowering it to 20% of 2a (entry 3). Furthermore, changing from dichloromethane to acetone resulted in the major formation of the side-product 3 (entry 4). Lower temperatures or slow addition of NMO did not provide any benefit to the reaction outcome (entries 5 and 6). The side-product 3 is presumably formed during the complexation step of alkyne 2a with the cobalt carbonyl complex. In order to shed light on the origin of 3, we synthesized the hexacarbonylic complex 411 to test it in the PKR. Adduct 2a was obtained in comparable yields to the one of the optimized conditions (compare entry 1 with 7). It is plausible to think that 1a forms two hexacarbonylic complexes, namely complex 4 and the corresponding standard hexacarbonylic complex bearing the terminal (deborylated)phenylacetylene, in which both undergo PKR forming 2a and 3, respectively. Experiments based on the decomposition of 2a by treatment with silica-gel or activated charcoal did not provide any traces of 3.
|
|
|||
|---|---|---|---|
| Entry | Change from optimized conditions | 2a (%) | 3 (%) |
| a 1a (0.6 mmol), Co2(CO)8 (1 equiv.), NBD (3 equiv.), and NMO (6 equiv.) in DCM (0.03 M) at r.t. for 16 h. b NMR yield using 1,1,2,2-tetrachloroethane as an internal standard. c Isolated yield. d Thermal conditions: 1a, Co2(CO)8 (1 equiv.), NBD (3 equiv.) in toluene (0.1 M) at 70 °C. e At 100 °C. f Via syringe pump. g 4 (0.2 mmol), NBD (3 equiv.), NMO (6 equiv.) in DCM (0.03 M) at r.t. | |||
| 1 | None | 89 (75)c | 9 |
| 2d | Using thermal conditions | 38 | 12 |
| 3d | Using thermal conditionse with 1 equiv. PPh3 | 20 | 13 |
| 4 | With acetone instead of DCM | 23 | 37 |
| 5 | 0 °C instead of r.t. | 70 | 15 |
| 6 | With slow addition of NMOf | 75 | 20 |
| 7 | Using complex 4g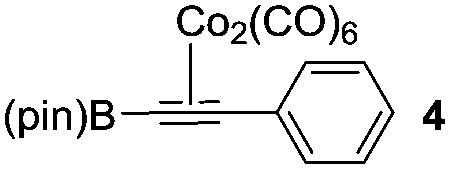 |
82 (75)c | 6 |

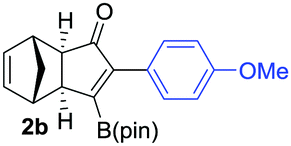
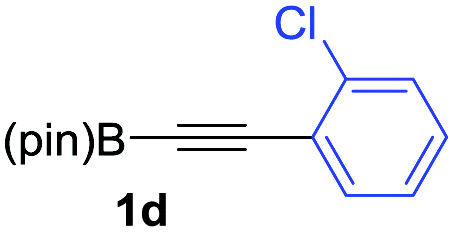
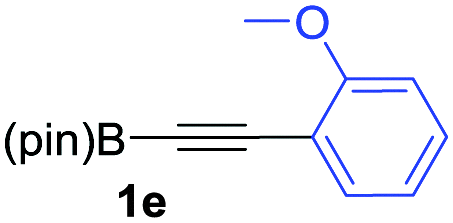
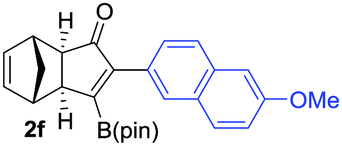
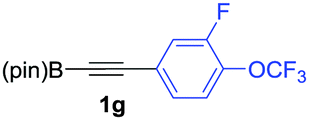
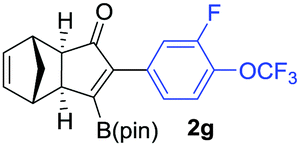
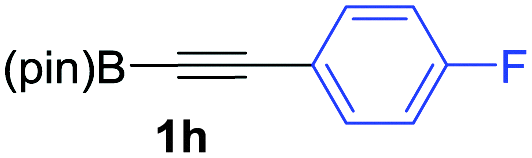
We next turned our attention to study the preparative scope of this reaction (Table 2). Towards this end, we succeeded to convert a wide range of terminal alkynes into their corresponding alkynylboronic pinacol esters (1a–1o), using the methodology developed by Brown and Srebnik.12 The fifteen internal alkynes reacted with norbornadiene, in the presence of Co2(CO)8 and NMO, to give a unique single exo-stereoisomer and the β-regioisomer of the α,β-substituted cyclopentenones (Table 2).
|
|
||||
|---|---|---|---|---|
| Entry | Alkynes 1 | Yield 1 (%) | PK adduct 2 | Yield 2 (%) |
| a As for Table 1, entry 1. b Isolated yields. c See the ESI for detailed synthetic procedure. d bzpin: benzopinacol. e dan: 1,8-diaminonaphthalene. | ||||
| 1 |

|
72 |
|
75 |
| 2 |

|
64 |
|
53 |
| 3 |
|
61 |
|
60 |
| 4 |
|
67 |
|
66 |
| 5 |
|
67 |

|
68 |
| 6 |

|
72 |
|
65 |
| 7 |
|
75 |
|
55 |
| 8 |
|
81 |

|
65 |
| 9 |

|
69 |
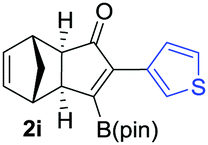
|
71 |
| 10 |

|
78 |

|
59 |
| 11 |

|
69 |
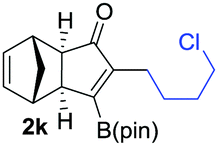
|
64 |
| 12 |

|
64 |

|
72 |
| 13 |

|
89 |
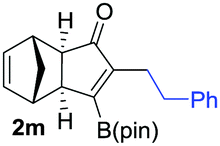
|
68 |
| 14c,d |
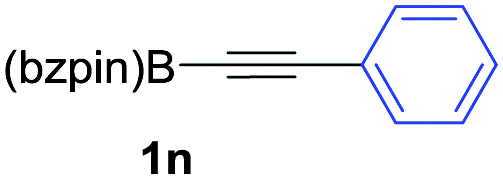
|
64 |
|
61 |
| 15c,e |

|
53 |
|
81 |

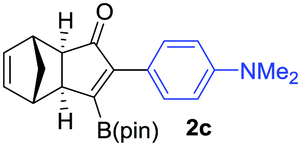
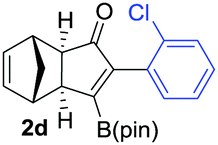
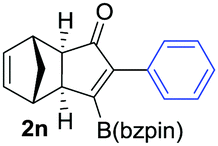
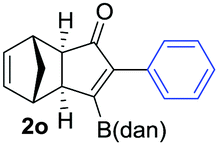
As shown in Table 2, the substitution pattern includes aromatic groups with a vast array of substituents with different electronic properties (entries 1–8), heteroaryl group (entry 9), olefinic (entry 10) or aliphatic groups (entries 11–13). However, when hindered alkynes such as tert-butylalkynylboronic pinacol ester or trimethylsilylalkynylboronic pinacol ester were used, the PKR failed to yield the corresponding adduct. Alkynes bearing Bbzpin (bzpin: benzopinacol) or Bdan (dan: 1,8-diaminonaphthalene) moieties were also tolerated (entries 14 and 15). In all cases, the resulting adducts were obtained with total stereo- and regioselectivity since none of the other possible isomers were detected by analytical means.13 Suitable crystals of 2a and 2o for X-ray diffraction analysis were successfully obtained for the first time from pentane and the selectivity of the pinacolboryl moiety at the β position of cyclopentenones could be unequivocally confirmed (Fig. 1). It confirms that the total regioselectivity observed is overweighed by electronic effects over the steric ones since adducts 2a and 2o, both bearing different steric requirements, exclusively placed the boryl moiety at the β position. Overall, we believe that the results presented in Table 2 not only demonstrate a high reactivity and functional group compatibility, but also the potential of alkynylboronic esters in the stereo- and regioselective PKR, comparable to the [2+2+1] cycloaddition from biased nonsymmetric internal alkynes,14 and the two-step protocol to form β-substituted cyclopentenones using the trifluoromethyl group as a removable steering group.15
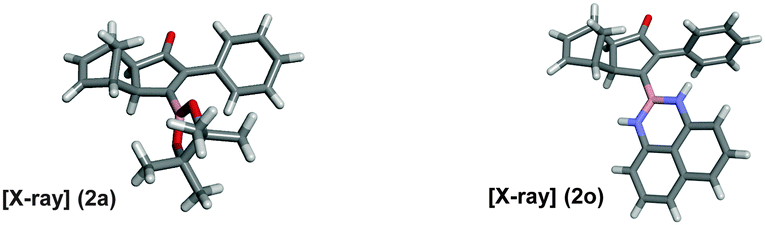 | ||
| Fig. 1 X-ray structures of compounds 2a and 2o. | ||
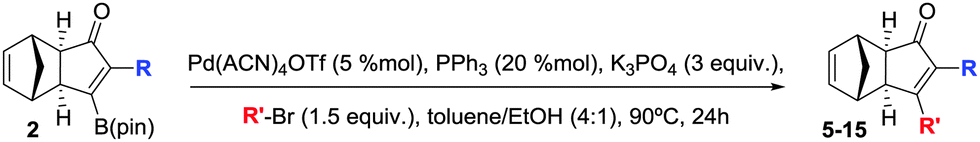
In order to demonstrate the usefulness of the β-boryl α-substituted cyclopentenones, we next turned our attention to their application towards the SMC, in line with our tunable synthetic strategy (Scheme 2) as a reliable and straightforward methodology.16 The reaction conditions were optimized using adduct 2a as a model substrate and 4-methoxyphenyl bromide as a reagent to furnish the desired α,β-disubstituted cyclopentenone 5 in 80% isolated yield (Table 3), in the presence of Pd(ACN)4OTf (5 mol%), PPh3 (20 mol%), and K3PO4 (3 equiv.) in a mixture of toluene/ethanol (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 90 °C during 24 hours. Our protocol was optimal for the assembly of several compounds regardless of their electronic properties or substitution patterns, providing the selective α,β-disubstituted cyclopentenones (5–15) with moderate to high isolated yields ranging 65–85% (Table 3). More interestingly, our methodology enabled the regioselective construction of challenging substrates that cannot be accessed through a standard PKR, for instances: (1) compounds 6, 7 and 10–12 were synthesized by installing the most hindered substituent at the β position, (2) compounds 5 and 7–9 bearing electronically or sterically non-differentiable substituents in both positions were successfully obtained, (3) isomers that are usually not accessible when carrying out the standard PKR such in the case of compound 12, holding an alkyl group at the α position. It should be highlighted that a number of heterocycles could be easily made. Crystals of 6 were successfully analysed by X-ray diffraction confirming the regioselectivity of the overall strategy claimed here.17 However, the strength of this methodology lies in the possibility of preparing specific regioisomers bearing negligible distinction in the substitution between α and β positions which would yield no selectivity through a standard PKR. Thus, compounds 14 and 15, containing minimal differentiation between both positions, such as phenyl versus d5-phenyl or 3-thionyl versus 3-furyl, could be successfully prepared.
:1) at 90 °C during 24 hours. Our protocol was optimal for the assembly of several compounds regardless of their electronic properties or substitution patterns, providing the selective α,β-disubstituted cyclopentenones (5–15) with moderate to high isolated yields ranging 65–85% (Table 3). More interestingly, our methodology enabled the regioselective construction of challenging substrates that cannot be accessed through a standard PKR, for instances: (1) compounds 6, 7 and 10–12 were synthesized by installing the most hindered substituent at the β position, (2) compounds 5 and 7–9 bearing electronically or sterically non-differentiable substituents in both positions were successfully obtained, (3) isomers that are usually not accessible when carrying out the standard PKR such in the case of compound 12, holding an alkyl group at the α position. It should be highlighted that a number of heterocycles could be easily made. Crystals of 6 were successfully analysed by X-ray diffraction confirming the regioselectivity of the overall strategy claimed here.17 However, the strength of this methodology lies in the possibility of preparing specific regioisomers bearing negligible distinction in the substitution between α and β positions which would yield no selectivity through a standard PKR. Thus, compounds 14 and 15, containing minimal differentiation between both positions, such as phenyl versus d5-phenyl or 3-thionyl versus 3-furyl, could be successfully prepared.
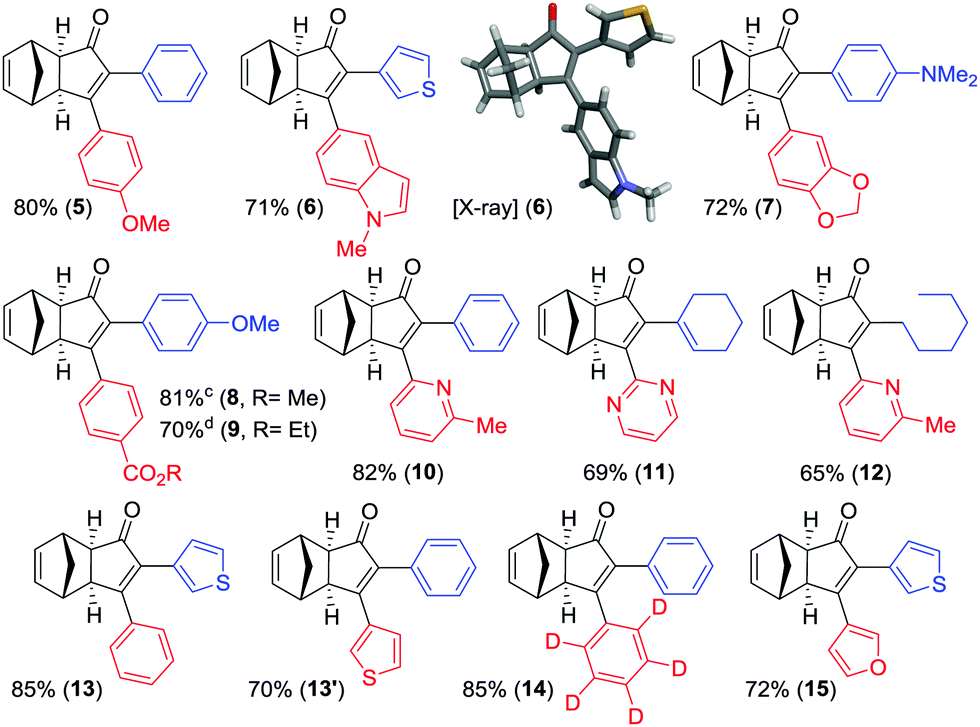
In order to better illustrate the potential of this strategy, we selected a few examples from the literature, which suffer from regioselectivity issues when synthesized through a standard PKR (Fig. 2). Direct comparison with representative closely-related compounds showed an substantial improvement in regioselectivity. Compounds 5 and 8 were obtained as a single regioisomer while a standard methodology afforded mixtures that are usually difficult to handle. Finally, cyclopentenones 13 and 13′ were independently obtained by simple reversal in the order of addition of the reagents, thus illustrating the ability to access both regioisomers.
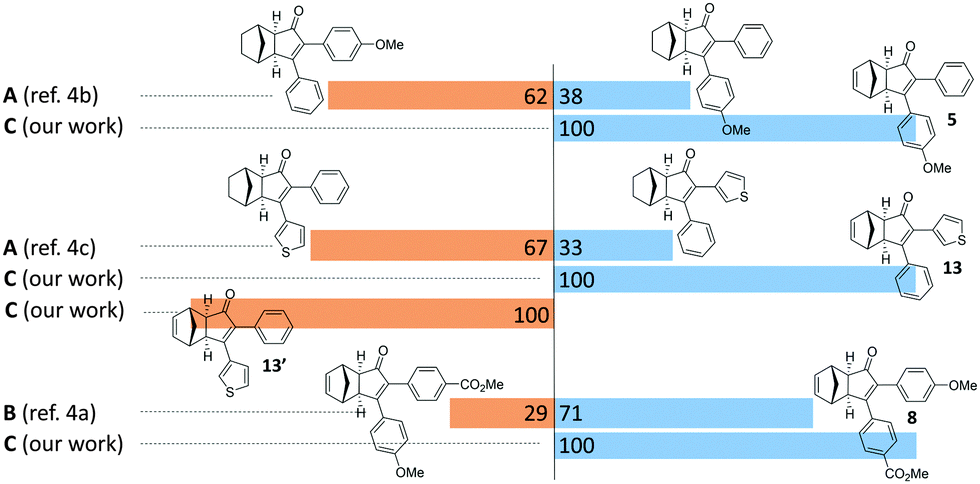 | ||
| Fig. 2 Regioselective outcome comparison of selected examples towards the application of our strategy. Reaction conditions: Method A: (a) Co2(CO)8, DCE; (b) norbornene, microwave, 90 °C. Method B: (a) Co2(CO)8, hexane; (b) norbornadiene, 60 °C. Method C: (a) as for Table 2; (b) as for Table 3. | ||
We eventually focus our strategy to the synthesis of a known scaffold of cyclooxygenase-2 (COX-2)18 (Scheme 3). COX-2 inhibitors are a type of non-steroidal anti-inflammatory drug that directly targets COX-2 enzymes, which are responsible for inflammation and pain. We tackled its synthesis using our sequential strategy starting from the alkynylboronic pinacol esters 1h. PKR afforded exclusively the regioisomer 2h (Table 2, entry 8), which was submitted to the SMC reaction with 4-bromophenyl methyl sulfone, yielding 16 in 88% isolated yield as a key scaffold.
 | ||
| Scheme 3 Synthesis of a key scaffold 16. | ||
In summary, we have described an intermolecular Pauson–Khand reaction conducted using internal alkynylboronic esters installing the boronic ester moiety at the β-position of the cyclopentenone with total regio- and stereoselectivity. More interestingly, the resulting adduct can be subsequently derivatised by Suzuki–Miyaura cross-coupling reaction allowing any combination of α,β-disubstituted cyclopentenones, solving a well-known and long-standing drawback about regioselective Pauson–Khand reactions. Overall, our approach also complements existing PKR protocols providing access to regioisomers that are not formed as the major isomers in the standard PKR. Currently, the methodology is being exploited using other challenging substrates.
We thank the Spanish Ministerio de Economia y Competividad (CTQ2013-43395-P) for financial support. T. L. thanks ACCIÓ for the TecnioSpring funding (TECSPR13-1-0040).
Notes and references
- I. U. Khand, G. R. Knox, P. L. Pauson, W. E. Watts and M. I. Foreman, J. Chem. Soc., Perkin Trans. 1, 1973, 977 RSC.
- Selected reviews: (a) The Pauson–Khand Reaction, ed. R. Ríos, Wiley-VCH, Weinheim, Germany, 2012 Search PubMed; (b) J. Blanco-Urgoiti, L. Anorbe, L. Perez-Serrano, G. Dominguez and L. Perez-Castells, Chem. Soc. Rev., 2004, 33, 32 RSC; (c) S. E. (née Thomas) Gibson and A. Stevenazzi, Angew. Chem., Int. Ed., 2003, 42, 1800 CrossRef PubMed; (d) K. M. Brummond and J. L. Kent, Tetrahedron, 2000, 56, 3263 CrossRef CAS; (e) Y. Keun Chung, Coord. Chem. Rev., 1999, 188, 297 CrossRef CAS.
- For examples of palladium-catalyzed PKR with moderated reverse of regioselectivity, see: N. Wu, L. Deng, L. Liu, Q. Liu, C. Li and Z. Yang, Chem. – Asian J., 2013, 8, 65 CrossRef CAS PubMed.
- Selected examples of regioselectivity issues, see: (a) Y. Ji, X. Verdaguer and A. Riera, Chem. – Eur. J., 2011, 17, 3942 CrossRef CAS PubMed; (b) B. E. Moulton, A. C. Whitwood, A. K. Duhme-Klair, J. M. Lynam and I. J. S. Fairlamb, J. Org. Chem., 2011, 76, 532 CrossRef PubMed; (c) A. Becheanu and S. Laschat, Synlett, 2002, 186 Search PubMed; (d) E. Fager-Jokela, E. Kaasalainen, K. Leppänen, J. Tois and J. Helaja, Tetrahedron, 2008, 64, 10381 CrossRef CAS.
- (a) E. Fager-Jokela, M. Muuronen, M. Patzschke and J. Helaja, J. Org. Chem., 2012, 77, 913 CrossRef PubMed; (b) E. Fager-Jokela, M. Muuronen, H. Khaizourane, A. Vázquez-Romero, X. Verdaguer, A. Riera and J. Helaja, J. Org. Chem., 2014, 79, 10999 CrossRef CAS PubMed.
- For an example of Nazarov with two different aryl groups appended, see: M. Wang, F. Han, H. Yuan and Q. Liu, Chem. Commun., 2010, 46, 2247 RSC.
- For selected examples of nickel- and palladium-catalyzed annulations, see: (a) J. Feng, G. Lu, M. Lv and C. Cai, J. Org. Chem., 2014, 79, 1056 Search PubMed; (b) M. L. N. Rao and R. J. Dhanorkar, Tetrahedron, 2014, 70, 806 Search PubMed; (c) Y. Shimamoto, H. Sunaba, N. Ishida and M. Murakami, Eur. J. Org. Chem., 2013, 142 Search PubMed; (d) J. Barluenga, P. Barrio, L. Riesgo, L. A. López and M. Tomás, J. Am. Chem. Soc., 2007, 129, 14422 CrossRef CAS PubMed.
- For instance, see: S. Suárez-Pantiga, E. Rubio, C. Alvarez-Rúa and J. M. González, Org. Lett., 2009, 11, 13 CrossRef PubMed.
- For selected examples of Suzuki–Miyaura cross-coupling reactions of alkynylboronic ester, see: (a) D. Ogawa, J. Li, M. Suetsugu, J. Jiao, M. Iwasaki and Y. Nishihara, Tetrahedron Lett., 2013, 54, 51 CrossRef; (b) A. I. Khalaf, J. K. Huggan, C. J. Suckling, C. L. Gibson, K. Stewart, F. Giordani, M. P. Barett, P. Wong, K. Barrack and W. N. Hunter, J. Med. Chem., 2014, 57, 6479 CrossRef CAS PubMed.
- For instances of cycloaddition reactions with alkynylboronic ester, see: (a) V. Gandon, D. Leca, T. Aechtner, K. P. C. Vollhardt, M. Malacria and C. Aubert, Org. Lett., 2004, 6, 340 CrossRef PubMed; (b) A. Geny, D. Leboeuf, G. Rouquié, K. P. C. Vollhardt, M. Malacria and C. Aubert, Chem. – Eur. J., 2007, 13, 540 CrossRef PubMed; (c) L. Iannazzo, K. P. C. Vollhardt, M. Malacria, C. Aubert and V. Gandon, Eur. J. Org. Chem., 2011, 3283 CrossRef CAS.
- Hexacarbonylic complexes bearing alkynylboronic ester have been already described and used in cyclooligomerization reactions in: A. Goswami, C.-J. Maier, H. Pritzkow and W. Siebert, Eur. J. Inorg. Chem., 2004, 2635 CrossRef CAS.
- H. C. Brown, N. G. Bhat and M. Srebnik, Tetrahedron Lett., 1988, 29, 2631 CrossRef CAS.
- Regioselectivity of an adduct of type 2 was confirmed by treating an analytical sample with TBAF in hot toluene yielding the corresponding protodeborylated product of type 3 which were compared with an authentic sample. For protocol, see: S. Nave, R. P. Sonawane, T. G. Elford and V. K. Aggarwal, J. Am. Chem. Soc., 2010, 132, 17096 CrossRef CAS PubMed.
- For an example of regioselective PKR using biased internal alkyne, see: A. Vázquez-Romero, L. Cárdenas, E. Blasi, X. Verdaguer and A. Riera, Org. Lett., 2009, 11, 3104 CrossRef PubMed.
- (a) N. Aiguabella, C. del Pozo, X. Verdaguer, S. Fustero and A. Riera, Angew. Chem., Int. Ed., 2013, 52, 535 Search PubMed; (b) N. Aiguabella, E. M. Arce, C. del Pozo, X. Verdaguer and A. Riera, Molecules, 2014, 19, 1763 CrossRef PubMed.
- For an alternative synthesis of unsymmetrical α,β-disubstituted cyclopentenones, a Heck reaction was used starting from the α-substituted adduct, see: (a) M. K. Gurjar, R. D. Wakharkar, A. T. Singh, M. Jaggi, H. B. Borate, P. D. Shinde, R. Verma, P. Rajendran, S. Dutt, G. Singh, V. K. Sanna, M. K. Singh, S. K. Srivastava, V. A. Mahajan, V. H. Jadhav, K. Dutta, K. Krishnan, A. Chaudhary, S. K. Agarwal, R. Mukherjee and A. C. Burman, J. Med. Chem., 2007, 50, 174 CrossRef PubMed; (b) M. K. Gurjar et al. , US Pat., Appl. 20030229146, 2003 Search PubMed.
- Compound 6 crystallizes in a 60:40 mixture corresponding to a rotation on the C–C bond between the thiophene moiety and the cyclopentenone core. For more details see CIF file.
- W. C. Black, C. Brideau, C.-C. Chan, S. Charleson, N. Chauret, D. Claveau, D. Ethier, R. Gordon, G. Greig, J. Guay, G. Hughes, P. Jolicoeur, Y. Leblanc, D. Nicoll-Griffith, N. Ouimet, D. Riendeau, D. Visco, Z. Wang, L. Xu and P. Prasit, J. Med. Chem., 1999, 42, 1274 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1483204–1483206. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc04717c |
| This journal is © The Royal Society of Chemistry 2016 |