
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural and mechanistic insights into a Bacteroides vulgatus retaining N-acetyl-β-galactosaminidase that uses neighbouring group participation†
C.
Roth
a,
M.
Petricevic
b,
A.
John
cd,
E. D.
Goddard-Borger
*cd,
G. J.
Davies
*a and
S. J.
Williams
*b
aDepartment of Chemistry, University of York, Heslington, York, UK. E-mail: gideon.davies@york.ac.uk
bSchool of Chemistry and Bio21 Molecular Science and Biotechnology Institute, University of Melbourne, Parkville, Victoria, Australia. E-mail: sjwill@unimelb.edu.au
cACRF Chemical Biology Division, The Walter and Eliza Hall Institute of Medical Research, Parkville, Victoria, Australia. E-mail: goddard-borger.e@wehi.edu.au
dDepartment of Medical Biology, University of Melbourne, Parkville, Victoria, Australia
First published on 15th August 2016
Abstract
Bacteroides vulgatus is a member of the human microbiota whose abundance is increased in patients with Crohn's disease. We show that a B. vulgatus glycoside hydrolase from the carbohydrate active enzyme family GH123, BvGH123, is an N-acetyl-β-galactosaminidase that acts with retention of stereochemistry, and, through a 3-D structure in complex with Gal-thiazoline, provide evidence in support of a neighbouring group participation mechanism.
The human gastrointestinal tract undergoes a constant process of renewal, leading to production of 5 × 107 cells per minute and the discharge of 250 g of cells per day.1 Colonocytes derived from the epithelia of the large intestine can be isolated at concentrations of up to 107 per gram of wet fecal material,2 and are an important nutrient source for the gut microbiota. A wide range of carbohydrates are represented on intestinal epithelial cells, including polysaccharides, glycoproteins and glycolipids. B. vulgatus is a widespread member of the human microbiota, and is generally considered beneficial, although it is an occasional opportunistic pathogen. B. vulgatus is typically present at levels of around 4% of bacteria in the gut microbiota, but can rise to constitute >40% of the gut microflora in Crohn's disease patients.3B. vulgatus, like other Bacteroides spp., possesses specialized polysaccharide utilization loci (PULs) encoding complex enzymatic machineries able to degrade and harness a wide variety of complex dietary polysaccharides along with SusC/SusD orthologues capable of importing oligosaccharide fragments into the periplasm.4 In addition, B. vulgatus and other members of the Bacteroidetes have specialised PULs for the degradation of intestinal mucosal glycoproteins and the glycocalyx.5 However, it appears that the ability of Bacteroides spp. to degrade host glycoconjugates is also enabled by enzymes encoded outside of the PULs. B. vulgatus encodes a single copy of an enzyme from glycoside hydrolase family GH123;9 a poorly characterized family with an emerging ability to degrade host-derived glycoconjugates. In this work we demonstrate that previously uncharacterized protein BvGH123 is a N-acetyl-β-galactosaminidase. We experimentally demonstrate for the first time that a member of this family hydrolyses glycosides with retention of anomeric stereochemistry, and provide evidence for a neighbouring group participation mechanism through an X-ray crystal structure in complex with an inhibitor that mimics the proposed intermediate of the reaction mechanism.
Previous studies of GH123 members, initially NagP from Paenibacillus sp.,6 and CpNga123 from C. perfringens (whose structure was recently solved)7 have led to the proposal of a two-step neighbouring group participation mechanism for catalysis (Fig. 1). According to such a mechanism, the enzyme possesses two key active site residues acting in the role of acid/base and transition state stabilization.8 In the first step, nucleophilic attack by the 2-acetamido group oxygen on the anomeric centre, assisted by charge stabilization and orientation by the transition state stabilizing residue, occurs simultaneously with protonation of the glycosidic oxygen by the acid/base residue to assist leaving group departure, resulting in the formation of an oxazolinium ion. In the second step, the acid/base residue acts as a base to assist in deprotonation of a nucleophilic water molecule, while the stabilizing residue facilitates ring-opening of the oxazolinium ion intermediate.9 Evidence in support of this mechanism was initially based on the nanomolar inhibition of NagP by Gal-thiazoline;6 an inhibitor that mimics the proposed oxazolinium ion intermediate (more formally a closely-related transition-state),10 along with the proposal of a catalytic diad of adjacent carboxyl residues reminiscent of those seen in β-hexosaminidases of GH20 and GH84, which are known to act through a neighbouring group participation mechanism.11,12 Recently, the first X-ray structure of a GH123 enzyme, CpNga123, in a product complex and a pseudo-Michaelis complex of an inactive mutant with substrate confirmed the identity of the catalytic diad, and revealed binding orientations of the 2-acetamido group that while not oriented directly below C1, was positioned in a manner consistent with that needed for neighbouring group participation.7
 | ||
| Fig. 1 Neighbouring group participation mechanism by a retaining N-acetyl-β-hexosaminidase. Hydroxyl groups have been omitted for clarity. | ||
To further investigate the mechanism of the GH123 galactosaminidases, residues 20–582 of ABR39589.1, encoding BVU_2198, without its signal peptide (hereafter BvGH123) was expressed in E. coli (Fig. S1, ESI†). Size exclusion chromatography-multi-angle laser light scattering revealed BvGH123 to be a dimer in solution. The enzyme displays a pH optimum of 5 using 4-nitrophenyl N-acetyl-β-D-galactosaminide (PNPGalNAc, 1) as a substrate (Fig. S2a, ESI†). The kinetic preference for β-galactosaminides versus β-glucosaminides was determined using PNPGalNAc and 4-nitrophenyl N-acetyl-β-D-glucosaminide (PNPGlcNAc). For PNPGalNAc, kcat/KM = 8.0 ± 0.4 × 103 M−1 s−1 and for PNPGlcNAc, kcat/KM = 3.8 ± 0.6 × 102 M−1 s−1; saturation could not be achieved, preventing the accurate determination of kcat and KM values. These data indicate a 21-fold preference for β-galactosaminides, consistent with previous reports on NagP6 and CpNga123,7 providing further evidence that the enzymes of this family are dedicated N-acetyl-β-galactosaminidases.
Previous studies have only speculated on the stereochemical outcome of substrate hydrolysis, which is of obvious importance for proposal of a feasible enzymatic mechanism.13 We used 1H NMR spectroscopy to monitor the stereochemical outcome of the enzyme-catalyzed hydrolysis of PNPGalNAc (1) (Fig. 2). Upon addition of enzyme, rapid consumption of substrate led to the production of a new product, with an anomeric signal at δ 4.63 ppm that displayed a large (J = 8 Hz) coupling constant. Over time, mutarotation led to the formation of a mixture of the first compound, and a new compound with an anomeric signal at δ 5.22 ppm characterized by a small (J = 4 Hz) coupling constant. These experimental data are consistent with the initial formation of β-GalNAc (2β), revealing that BvGH123, and by inference all GH123 members, act with a net retention of anomeric configuration. Using isothermal titration calorimetry, Gal-thiazoline14 was shown to bind BvGH123 with KD = 9.6 nM (Fig. S2b, ESI†). Collectively, these results support previous suggestions that enzymes of family GH123 use a neighbouring group participation mechanism involving the 2-acetamido group.
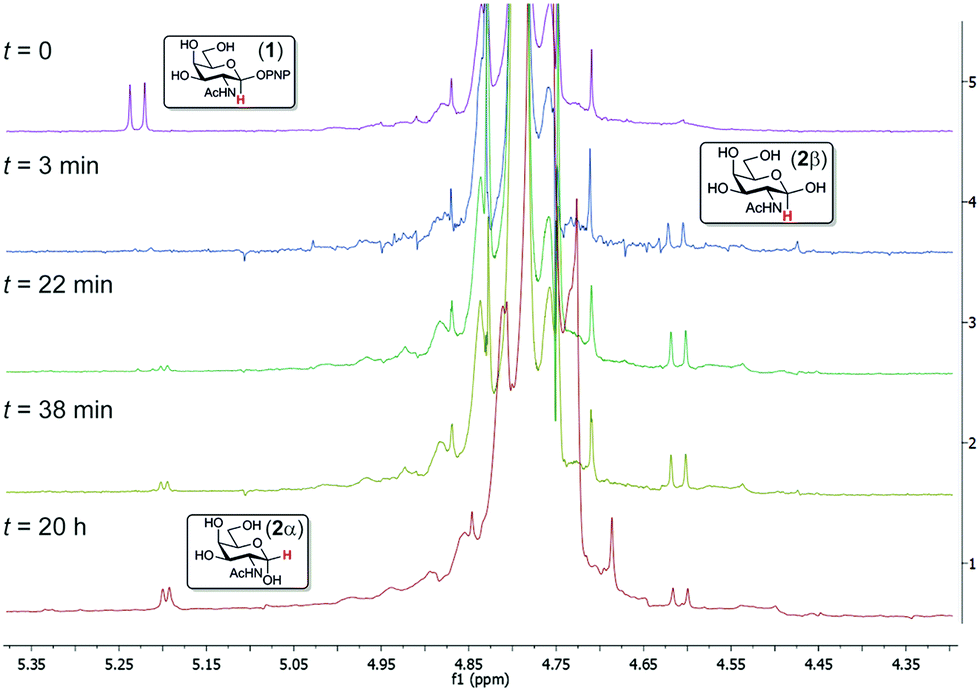 | ||
| Fig. 2 Stacked 1H NMR spectra showing time course of BvGH123 hydrolysis of PNPGalNAc. Substrate hydrolysis results in initial release of β-GalNAc with retention of anomeric configuration, which then undergoes mutarotation. | ||
At the time of our structure solution, no GH123 structure had been reported and thus the three-dimensional structure of BvGH123 was solved by experimental phasing using single anomalous diffraction from a selenomethionine-substituted derivative and subsequently refined against a native dataset to a resolution of 1.85 Å (Table 1 and Table S1, ESI†). The overall structure reveals a two domain structure with an N-terminal β-sheet domain (residues 1–195) followed by a catalytic (β/α)8-barrel domain (residues 196–563) (Fig. 3a). The latter has three insertions forming a 4-stranded β-wing domain and a C-terminal α-helical cap, as observed recently in the CpGH123 structure.7 Structures of BvGH123 and CpNga123 overlap with an r.m.s.d. of approximately 1.7 Å over 500 residues. Differences include a longer N-terminus for BvGH123, and a C-terminal β-strand insertion followed by a longer C-terminal helix for CpNg123. Additionally a disulfide bond joining Cys117 and Cys430 is observed only in BvGH123, connecting the loop comprising residues 110–120 with the 430-loop which is part of the active site. Sequence conservation within the GH123 family is relatively low and is mainly confined to the catalytic residues and sections of the (β/α)8-barrel. The N-terminal β-sheet domain, found in both structures, shows weak structural similarity to the IgG-fold found in several antigens and adhesion modules, as well as within Titin, and might facilitate adhesion to the cell surface of the host, helping to degrade cell surface glycans.15a Remote structural similarity is also observed for the BACON-domain found, for example, in GH5 enzymes.5 A stable assembly in the crystal lattice identified by PISA16 is formed by interaction of the 4-stranded β-wing domain formed by insertions between the second and third strands and the helix of the (β/α)8-barrel (Fig. S3, ESI†), presumably representing the dimer observed in solution.
 | ||
| Fig. 3 Structures of Bacteroides vulgatus GH123 and complexes with Gal-thiazoline and GalNAc. (a) Overview of the BvGH123 structure in complex with Gal-thiazoline, rainbow-coloured from the N- to C-terminus; Gal-thiazoline is shown as space filled van der Waals surface. Views of the active site of BvGH123 in complex with (b) GalNAc and (c) Gal-thiazoline, highlighting the active site residues. The 2mFo − DFc map for GalNAc in blue is contoured at 0.22 e Å−3 (1σ r.m.s.d.) and 0.25 e Å−3 (1.5σ r.m.s.d.) for Gal-thiazoline. The C1–O interaction of the N-acetyl group in the GalNAc complex is highlighted with a red dotted line. (d) Interaction plot of Gal-thiazoline within the active site. | ||
| Native (apo) | BvGH123–GalNAc | BvGH123–Gal-thiazoline | |
|---|---|---|---|
| Space group | P212121 | P212121 | P212121 |
| Resolution limit | 1.85 Å | 2.1 Å | 2.3 Å |
| R merge | 0.141(1.930) | 0.104(1.530) | 0.063(1.552) |
| I/σI | 9.9(1.2) | 12.0(1.2) | 15.6(1.1) |
| Completeness (%) | 99.9(100) | 100(100) | 93.4(63.6) |
| R.m.s.d bond lengths (Å) | 0.016 | 0.014 | 0.013 |
| R.m.s.d bond angles (°) | 1.70 | 1.59 | 1.63 |
To gain insight into the structural basis of the retaining mechanism of BvGH123, we solved structures of complexes with the product GalNAc (Fig. 3b), as well as Gal-thiazoline at resolutions of 2.1 and 2.3 Å, respectively (Fig. 3c). Compared to the “apo” form, both complexes revealed a movement both of several of the loops lining the active site and of key residues, which is most pronounced for W306 and the adjacent pair of catalytic residues D361 and E362 (Fig. 4 and Fig. S4, ESI†). Similar movements were observed for CpNga123 upon engagement with carbohydrate.7 In the case of BvGH123 upon binding to Gal-thiazoline the loop containing D361 and E362 moves to engage these acidic side chains into a closed conformation in which the catalytic residues engage in close contacts with the inhibitor. As the inhibitor is a mimic of the oxazolinium intermediate (or possibly the transition state),15b these interactions appear to represent those of a catalytically competent form of the enzyme.
 | ||
| Fig. 4 BvGH123 undergoes conformational changes upon ligand binding. Comparison of the conformations for the active site residues in: (a) fully open form of apo BvGH123; (b) partially closed form in complex with GalNAc; (c) fully closed form in complex with Gal-thiazoline. | ||
The X-ray structures of the inhibitor and product complexes of BvGH123 provide an intimate view of the geometry of the active site. In the BvGH123–Gal-thiazoline complex, which is in a fully closed state, the adjacent carboxyl residues D361 and E362 adopt positions consistent with roles as transition state stabilizer and acid/base, respectively (Fig. 3c). The BvGH123–GalNAc product complex revealed only a partially closed conformation with W306 nestled above the pyranose and D361 engaging with the acetamido group of GalNAc (Fig. 3b). This complex contrasts with that reported for GalNAc bound to CpNga123, which is in an open conformation (Fig. S4, ESI†).7 The pyranose ring of Gal-thiazoline adopts a 4C1 conformation consistent with an expected 1S3 → 4H3 → 4C1 conformational itinerary for the glycosylation half-reaction.17 Complexes of CpNga123-D344N/E345Q with substrate (ganglioside glycans GA2 or Gb4) were observed in a 4C1 conformation, which is inconsistent with the substrate distortion expected with a pseudo-axial orientation of the anomeric group, as anticipated for stereoelectronic reasons.7 Combined with the location of the 2-acetamido group away from C1, the Michaelis complexes obtained with CpNga123 have been suggested to represent non-catalytically-competent complexes in which the open conformation prevents the active centre residues from fully engaging with the substrate.
A comparison of the active sites of GH families that are known to utilise a substrate-assisted retaining mechanism, which includes families 18, 20, 25, 84 and 85, reveal a remarkable spatial conservation of the catalytic residues located on the fourth β-strand of the respective (β/α)8-barrel (Fig. 5). Families GH20 and 84, like GH123, have the catalytic acid/base and stabilizer adjacent in their protein chains, and have essentially identical arrangements of these groups and the linking peptide backbone. Families GH18, 25 and 85 have a one residue insertion between the catalytic residues, yet still adopt a remarkably similar spatial arrangement. Likewise the active site closure motion now observed for two GH123 enzymes is also seen for a GH20 hexosaminidase and a GH84 O-GlcNAcase.7,12,22 These hexosaminidase families have little or no sequence conservation yet display similar folds and arrangement of catalytic residues and use a neighbouring group participation mechanism, suggesting a common solution to the chemical problem of hydrolysis of N-acetyl-β-hexosaminides.23
 | ||
| Fig. 5 Overlay of BvGH123–Gal-thiazoline active site residues (wheat) with members of other neighbouring group participation enzyme families (GH18, 20, 25, 84, 85). (a) Overlay of family representatives with adjacent catalytic residues: GH20 Paenibacillus sp. β-HexNAcase (purple, PDB: 3SUR),18 and GH84 Bacteroides thetaiotaomicron O-GlcNAcase (light green, PDB: 2CHN).11 (b) Overlay of family representatives with non-adjacent catalytic residues: GH18 Serratia marcescens chitinase A (grey, PDB: 2WK2),19 GH25 Aspergillus fumigatus lysozyme (steel blue, PDB: 2X8R),20 and GH85 Streptococcus pneumoniae endo-D (plum, PDB: 2W92).21 | ||
This work was supported by the Australian Research Council (FT130100103), and the BBSRC (BB/K003836/1), and the Ramaciotti Foundation and VESKI with additional support from the Australian Cancer Research Foundation and Victorian State Government Operational Infrastructure Support, NHMRC IRIISS grant 9000220. We thank the Diamond Light Source for access to beamlines I02, I03 and I04 (proposal number mx-9948) that contributed to the results presented here. We also thank Dr Andrew Leech at the Bioscience Technology Facility in the Department of Biology in York for his help with the SEC-MALLS experiments. We are grateful to Dr Johan Turkenburg and Sam Hart for assistance during data collection and Wendy Offen for help with the crystallisation.
Notes and references
- P. G. Falk, L. V. Hooper, T. Midtvedt and J. I. Gordon, Microbiol. Mol. Biol. Rev., 1998, 62, 1157 CAS.
- D. P. Bojanova and S. R. Bordenstein, PLoS Biol., 2016, 14, e1002503 Search PubMed.
- J. G. H. Ruseler-van Embden, R. van der Helm and L. M. C. van Lieshout, FEMS Microbiol. Lett., 1989, 58, 37 CrossRef CAS.
- (a) B. A. White, R. Lamed, E. A. Bayer and H. J. Flint, Annu. Rev. Microbiol., 2014, 68, 279 CrossRef CAS PubMed; (b) N. Terrapon, V. Lombard, H. J. Gilbert and B. Henrissat, Bioinformatics, 2015, 31, 647 CrossRef CAS PubMed; (c) J. Larsbrink, T. E. Rogers, G. R. Hemsworth, L. S. McKee, A. S. Tauzin, O. Spadiut, S. Klinter, N. A. Pudlo, K. Urs, N. M. Koropatkin, A. L. Creagh, C. A. Haynes, A. G. Kelly, S. N. Cederholm, G. J. Davies, E. C. Martens and H. Brumer, Nature, 2014, 506, 498 CrossRef CAS PubMed; (d) J. Larsbrink, A. J. Thompson, M. Lundqvist, J. G. Gardner, G. J. Davies and H. Brumer, Mol. Microbiol., 2014, 94, 418 CrossRef CAS PubMed; (e) G. R. Hemsworth, G. Dejean, G. J. Davies and H. Brumer, Biochem. Soc. Trans., 2016, 44, 94 CrossRef CAS PubMed.
- N. A. Pudlo, K. Urs, S. S. Kumar, J. B. German, D. A. Mills and E. C. Martens, mBio, 2015, 6, e01282 CrossRef PubMed.
- T. Sumida, K. Fujimoto and M. Ito, J. Biol. Chem., 2011, 286, 14065 CrossRef CAS PubMed.
- I. Noach, B. Pluvinage, C. Laurie, K. T. Abe, M. G. Alteen, D. J. Vocadlo and A. B. Boraston, J. Mol. Biol., 2016, 428, 3253 CrossRef CAS PubMed.
- D. J. Vocadlo and G. J. Davies, Curr. Opin. Chem. Biol., 2008, 12, 539 CrossRef CAS PubMed.
- S. J. Williams, B. Mark, D. J. Vocadlo, M. N. James and S. G. Withers, J. Biol. Chem., 2002, 277, 40055 CrossRef CAS PubMed.
- (a) M. S. Macauley, G. E. Whitworth, A. W. Debowski, D. Chin and D. J. Vocadlo, J. Biol. Chem., 2005, 280, 25313 CrossRef CAS PubMed; (b) G. E. Whitworth, M. S. Macauley, K. A. Stubbs, R. J. Dennis, E. J. Taylor, G. J. Davies, I. R. Greig and D. J. Vocadlo, J. Am. Chem. Soc., 2007, 129, 635 CrossRef CAS PubMed.
- R. J. Dennis, E. J. Taylor, M. S. Macauley, K. A. Stubbs, J. P. Turkenburg, S. J. Hart, G. N. Black, D. J. Vocadlo and G. J. Davies, Nat. Struct. Mol. Biol., 2006, 13, 365 CAS.
- M. Robb, C. S. Robb, M. A. Higgins, J. K. Hobbs, J. C. Paton and A. B. Boraston, J. Biol. Chem., 2015, 290, 30888 CrossRef CAS PubMed.
- (a) E. C. Lai and S. G. Withers, Biochemistry, 1994, 33, 14743 CrossRef CAS PubMed; (b) S. Drouillard, S. Armand, G. J. Davies, C. E. Vorgias and B. Henrissat, Biochem. J., 1997, 328, 945 CrossRef CAS PubMed; (c) B. L. Mark, D. J. Vocadlo, S. Knapp, B. L. Triggs-Raine, S. G. Withers and M. N. G. James, J. Biol. Chem., 2001, 276, 10330 CrossRef CAS PubMed.
- S. Knapp and D. S. Myers, J. Org. Chem., 2002, 67, 2995 CrossRef CAS PubMed.
- (a) F. Bäckhed, R. E. Ley, J. L. Sonnenburg, D. A. Peterson and J. I. Gordon, Science, 2005, 307, 1915 CrossRef PubMed; (b) N. Cekic, J. E. Heinonen, K. A. Stubbs, C. Roth, Y. He, A. J. Bennet, E. J. McEachern, G. J. Davies and D. J. Vocadlo, Chem. Sci., 2016, 7, 3742 RSC.
- E. Krissinel, Nucleic Acids Res., 2015, 43, W314 CrossRef PubMed.
- Y. He, M. S. Macauley, K. A. Stubbs, D. J. Vocadlo and G. J. Davies, J. Am. Chem. Soc., 2010, 132, 1807 CrossRef CAS PubMed.
- T. Sumida, K. A. Stubbs, M. Ito and S. Yokoyama, Org. Biomol. Chem., 2012, 10, 2607 CAS.
- J. M. Macdonald, C. A. Tarling, E. J. Taylor, R. J. Dennis, D. S. Myers, S. Knapp, G. J. Davies and S. G. Withers, Angew. Chem., Int. Ed., 2010, 49, 2599 CrossRef CAS PubMed.
- J. E. Korczynska, S. Danielsen, U. Schagerlof, J. P. Turkenburg, G. J. Davies, K. S. Wilson and E. J. Taylor, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2010, 66, 973 CrossRef CAS PubMed.
- D. W. Abbott, M. S. Macauley, D. J. Vocadlo and A. B. Boraston, J. Biol. Chem., 2009, 284, 11676 CrossRef CAS PubMed.
- J. F. Darby, J. Landstrom, C. Roth, Y. He, G. J. Davies and R. E. Hubbard, Angew. Chem., Int. Ed. Engl., 2014, 53, 13419 CrossRef CAS PubMed.
- R. R. Copley and P. Bork, J. Mol. Biol., 2000, 303, 627 CrossRef CAS PubMed; A. D. Goldman, J. T. Beatty and L. F. Landweber, J. Mol. Evol., 2016, 82, 17 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: SI figures, cloning, expression, biochemistry, and crystallography methods. See DOI: 10.1039/c6cc04649e |
| This journal is © The Royal Society of Chemistry 2016 |