DOI:
10.1039/C6CC04523E
(Feature Article)
Chem. Commun., 2016,
52, 10938-10947
Design directed self-assembly of donor–acceptor polymers
Received
30th May 2016
, Accepted 7th July 2016
First published on 7th July 2016
Abstract
Donor–acceptor polymers with an alternating array of donor and acceptor moieties have gained particular attention during recent years as active components of organic electronics. By implementation of suitable subunits within the conjugated backbone, these polymers can be made either electron-deficient or -rich. Additionally, their band gap and light absorption can be precisely tuned for improved light-harvesting in solar cells. On the other hand, the polymer design can also be modified to encode the desired supramolecular self-assembly in the solid-state that is essential for an unhindered transport of charge carriers. This review focuses on three major factors playing a role in the assembly of donor–acceptor polymers on surfaces which are (1) nature, geometry and substitution position of solubilizing alkyl side chains, (2) shape of the conjugated polymer defined by the backbone curvature, and (3) molecular weight which determines the conjugation length of the polymer. These factors adjust the fine balance between attractive and repulsive forces and ensure a close polymer packing important for an efficient charge hopping between neighboring chains. On the microscopic scale, an appropriate domain formation with a low density of structural defects in the solution deposited thin film is crucial for the charge transport. The charge carrier transport through such thin films is characterized by field-effect transistors as basic electronic elements.

Tomasz Marszalek (left), Mengmeng Li (middle) and Wojciech Pisula (right)
| Tomasz Marszalek has been a postdoctoral researcher in the Group of Professor Klaus Müllen at Max Planck Institute for Polymer Research since 2012. He completed his PhD under the supervision of Professor Jacek Ulanski in the Department of Molecular Physics at Lodz University of Technology. His research interests lie in the field of organic electronics and self-assembly of low and high molecular weight organic semiconductors. |
| Mengmeng Li obtained his PhD degree under the supervision of Professor Klaus Müllen at the department of synthetic chemistry of the Max Planck Institute for Polymer Research in Mainz in 2016. His main research interests include self-assembly control of organic semiconductors in field-effect transistors and other related electronic devices. |
| Wojciech Pisula studied chemical engineering at the University of Applied Science Osnabrück and at the University of Wales, Swansea, where he gained his Master of Science. In 2001, he joined the group of Professor Klaus Müllen at the Max Planck Institute for Polymer Research, where he completed his dissertation in 2005. In 2006 he changed to a full position at Evonik Industries AG keeping at the same time his status as project leader at the MPIP. At the beginning of 2015 he received his habilitation in material science at the Technical University of Darmstadt under the mentorship of Professor Heinz von Seggern. Since mid 2015 he has been additionally employed as an associate professor at the Lodz University of Technology in the Department of Molecular Physics of Professor Jacek Ulanski. Wojciech's research interests are physical chemical aspects of π-conjugated self-organizing systems and the resulting functionalities. |
1. Introduction
Organic electronics have found the way from basic academic research to industrial applications over recent years in a quickly growing market. This market covers especially application fields in light emitting diodes (LEDs),1,2 field-effect transistors (FETs)3,4 and solar cells5 which open the window for novel technologies. Organic semiconductors consist of conjugated molecules which can be processed into devices from solution, allowing a large surface-area.6,7 Moreover, many active compounds are mechanically flexible and therefore applicable to bendable electronic elements.8 Due to the low cost processing, one-way applications of electronic elements are realizable like radio frequency identification (RFID) tags and sensors in which FETs play a major role.9,10 The performance of a transistor is mainly determined by the speed of the charge carrier transport from one electrode to the other which in turn depends on various factors related to the semiconducting system which are discussed in the next paragraph in more detail.11–13
Organic semiconductors can consist of either small conjugated molecules or long polymers. In both cases, the π-conjugated bonds lead to delocalized filled and empty π-orbitals which affect the electrical and optical behavior of the semiconductor.14 The (macro)molecules weakly interact in solution and solid-state by π-stacking, van der Waals or dipole–dipole forces leading to so-called self-assembly into distinct aggregates and superstructures.15 This self-assembly of the polymer at the nano- and mesoscale determines the transport of charge carries through the active layer.16–18 Small molecules and polymers differ significantly in their assembly and transport properties. The first type of system shows a pronounced tendency to strongly crystallize leading to a high degree of (long-range) order and close π-stacking, defined here as packing. However, small molecules suffer from inhomogeneous film formation and create distinct grain boundaries as effective charge carrier traps due to their high crystallinity. Their additional apparent electronic anisotropy makes an implementation into large transistor arrays challenging. In contrast, conjugated polymers organize in layer structures that are less sensitive to the relative lattice orientation towards the transport direction.19 It is assumed that a fast charge carrier migration takes place along the conjugated backbone, while hopping of charges occurs between polymer chains preferentially along the π-stacking direction. When deposited on the device surface from solution, macromolecular semiconductors organize into complex and hierarchical superstructures over different length scales.20 A high performance device requires an optimization over a wide range from a few Å to several micrometers which is critical for the device. The above mentioned local packing in the range of a few Å to a few nm within a periodic lattice is dictated by the polymer design and chemistry and thus by intermolecular interactions. Locally, the relative polymer backbone arrangement has impact on the π-orbital overlap of neighboring chains and affects the charge hopping. In contrast to small crystalline molecules, conjugated polymers reveal also micrometer large areas of varying order leading to so-called semicrystalline microstructures with separated regions of both high crystallinity and disorder (amorphous). It is assumed that in polymers with ultrahigh charge carrier mobilities the ordered domains are interconnected by loose polymer chains that ensure a charge transfer.21 The nucleation and growth during the self-assembly process into corresponding structures is governed also by the polymer intermolecular interactions. At a meso- and macroscopic scale, crystallite size and texture become relevant structural parameters for the charge carrier transport. Long-range order, large domain size as well as crystallite orientation with respect to neighboring domains and transport direction favor the conduction in the device.22 Disordered regions and pronounced grain boundaries between small domains limit the performance of a transistor. Additionally, the polymer orientation with respect to the surface is another important factor for the device operation. The backbones can arrange either in a face- or edge-on fashion on the substrate. In an edge-on organization, the aromatic plane is normal to the surface so that the π-stacking axis is oriented parallel to the substrate. This orientation is required for application in transistors where the transport occurs also parallel to the surface close to the dielectric/semiconductor interface.
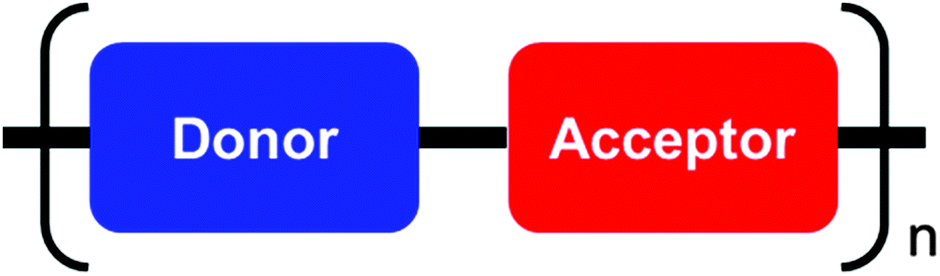
For a long time the charge carrier mobilities in polymer transistors have been generally low due to the poor packing and lack of macroscopic order of these materials.23 There have been several approaches towards enforcing thin film crystallinity in order to solve this problem.24 In contrast to homopolymers, donor–acceptor (D–A) copolymers consist of an alternating arrangement of units of different electron affinity and are considered as rigid-rod macromolecules (Fig. 1).25 The low band gap of such polymeric systems makes them attractive for applications in organic photovoltaic cells.26,27 Moreover, in many cases the planar arrangement of the conjugated backbone ensures a close π-stacking distance which is relevant for efficient intermolecular charge carrier transport.28 A close packing of the polymer chains might be favored from additional donor–acceptor interactions.
 |
| | Fig. 1 General structure of a simplest D–A polymer backbone consisting of an electron deficient and an electron rich unit. D–A polymers can also comprise more units within one monomeric building block. | |
This review discusses the role of the macromolecular structure of D–A copolymers on their local packing, supramolecular organization, and microstructure formation. Understanding the interplay between macromolecular design, organization of the polymer within nano- and microscopic dimensions as well as charge carrier transport opens the door towards the development of new polymeric semiconductors of superior performance for future applications. The structure variety in the discussed D–A polymers contains a broad toolbox of crucial structural features including molecular weight, bulkiness and substitution position of the alkyl chains and backbone curvature.
2. General features of donor–acceptor polymers
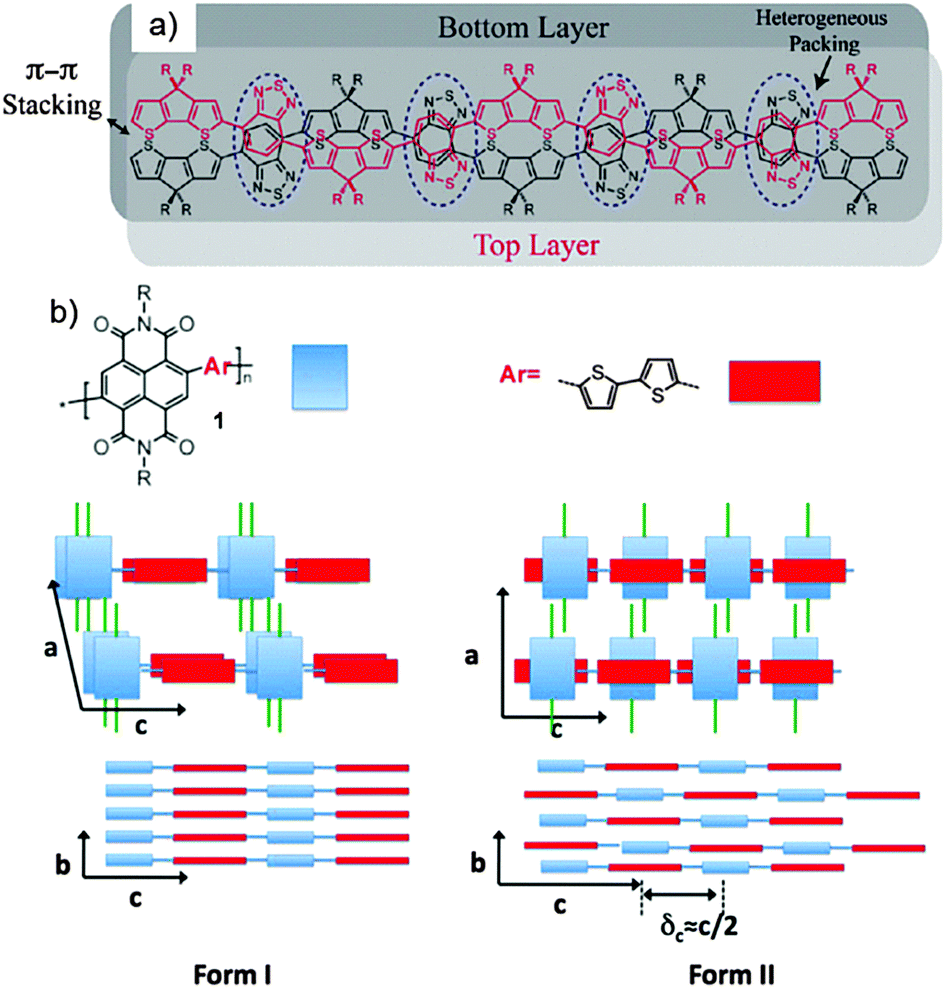
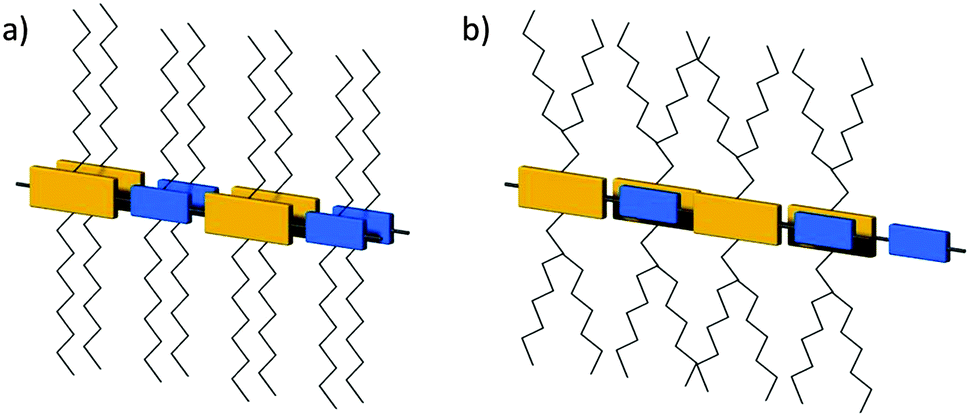
As said before, the backbone of this polymer type is built on one or more electron donors and electron acceptors (Fig. 1). The modification of the electron pushing and pulling strength of the units allows controlling of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels of the polymer. This is possible since the HOMO is mainly sited at the donor unit, while the LUMO is related to the acceptor. Consequently, the band gap can be reduced to even around 1 eV.29 This low value makes D–A polymers attractive for solar cell applications due to an increase of the absorption over a broad wavelength range. Recently, D–A polymers revealed in heterojunction solar cells in combination with fullerenes power conversion efficiencies up to 10%.30 The extended intrachain conjugation and intense donor–acceptor interactions via electron transfer lead to a small torsion angle favoring tight π-stacking packing of the polymer chains in the layer structure and high crystallinity. Consequently, D–A polymers with these characteristics show record mobilities in FETs far beyond 5 cm2 V−1 s−1 for holes and electrons.31,32 The intralayer stacking of the D–A polymers can be determined by either solid-state nuclear magnetic resonance (NMR) or electron diffraction. It was proven by a combination of solid-state NMR and X-ray diffraction (XRD) that the hexadecane substituted cyclopentadithiophene–benzothiadiazole polymer 2a is π-stacked in a lamellar kind of fashion with donor and acceptor units ordered in an alternating way (Fig. 2a).33
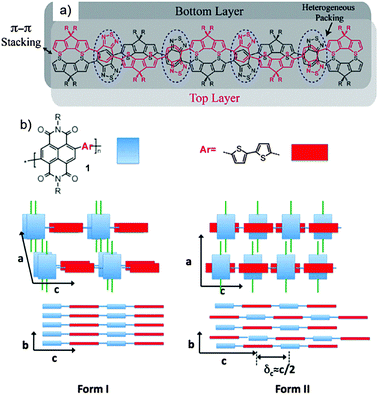 |
| | Fig. 2 Schematic drawing illustrating the local packing of donor–acceptor groups in two neighboring polymer chains of (a) 2a (blue dashed circles mark regions where the acceptor groups are heterogeneously packed on top of one another) and (b) of 1 with NDI and T2 units in forms I and II. In form I, NDI and T2 units are forming segregated columns, while in II, the units are in π-overlap one with another. Reprinted with permission from ref. 33 and 34, Copyright 2011 and 2012, American Chemical Society. | |
In this model, the acceptor groups in adjacent layers are located on top of each other. This seems to be an optimal polymer packing for 2a as the long hexadecane side chains avoid steric clash. For another type of D–A polymer, a naphthalene–diimide–bithiophene polymer 1, two distinct polymorphs have been found in oriented films depending on the underlining substrate.34 During directional epitaxial crystallization from 1,3,5-trichlorobenzene polymer 1 organizes in segregated columns of bithiophene (2T) and naphthalene diimide (NDI) units leading to form I (Fig. 2b). On poly(tetrafluoroethylene) (PTFE) substrates form II is obtained after cooling the film from the melt with c/2 shifted neighboring backbones so that NDI and T2 units are in π-overlap one with another (Fig. 2b). Form I can be transferred to form II by annealing at a temperature above 250 °C. Due to different π-stacking modes of NDI and T2, the two polymorphs show characteristic signatures in the UV-vis spectra. A similar change in packing mode was reported in spin-coated poly(octylfluorene-alt-benzothiadiazole) (3) films of high molecular weight (Fig. 3).35 The columnar type of segregated packing was related to the large torsion angle between the F8 and BT units decreasing the distance between neighboring chains.
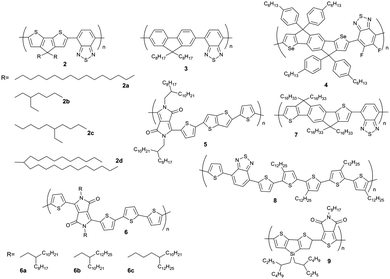 |
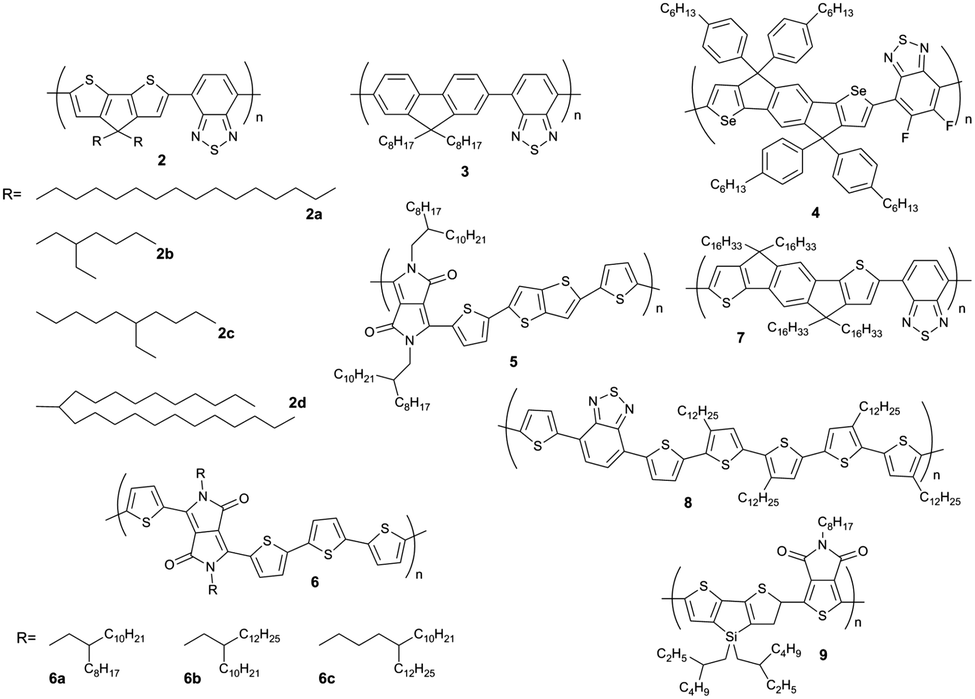
| | Fig. 3 Chemical structures of D–A polymers chosen to describe the influence of molecular weight on charge carrier mobility. | |
3. Molecular weight
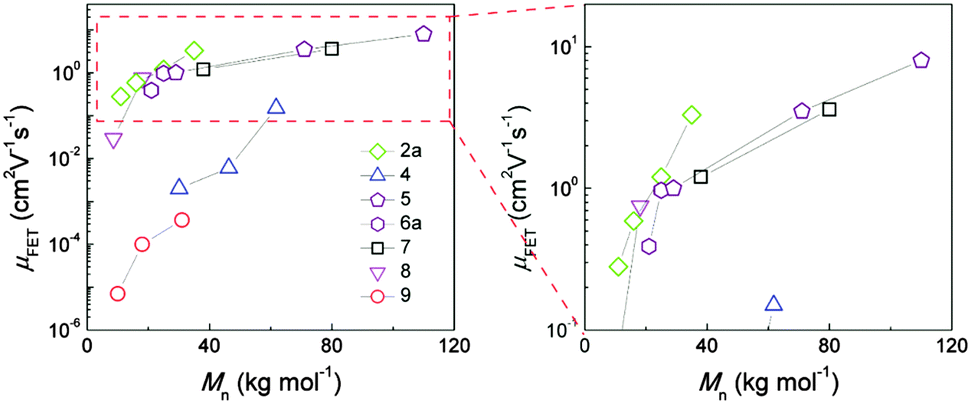
The molecular weight of D–A copolymers plays a critical role for their charge carrier transport. Fig. 4 summarizes the field-effect mobilities of several D–A polymers as a function of number average molecular weight Mn neglecting the polydispersity for simplification. A general trend can be identified with thin films of higher Mn polymers exhibiting larger mobilities. For instance, with doubling Mn from 30.1 to 61.8 kg mol−1, the average hole mobility of 4 (Fig. 3) can be enhanced by two orders of magnitude from 2 × 10−3 to 0.15 cm2 V−1 s−1.39 A 10-fold improvement in hole transport for 2 has been also observed when increasing Mn from 11 to 35 kg mol−1 yielding a maximum mobility of 3.3 cm2 V−1 s−1.33 In the case of a DPP–DTT polymer 5, ultrahigh molecular weight has been achieved with Mn = 110 kg mol−1 resulting in an extremely high mobility with an average of 8 cm2 V−1 s−1 and maximum of 10.5 cm2 V−1 s−1. On the contrary, the low molecular weight 5 (Mn = 29 kg mol−1) shows a much poorer transistor performance with a mobility of about 1 cm2 V−1 s−1.44
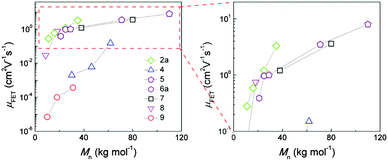 |
| | Fig. 4 Dependence of field-effect mobility of D–A polymers on molecular weight.36–43 | |
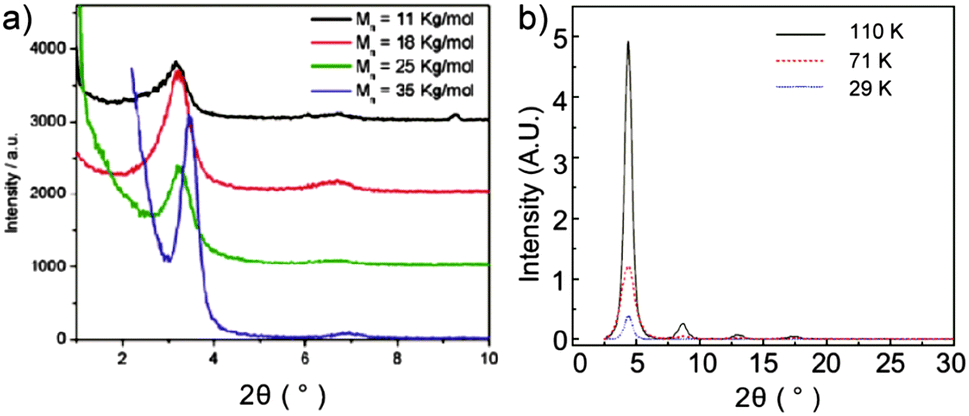
One explanation for the Mn dependent mobility is that ordered regions in films are more densely interconnected by longer polymer chains facilitating charge carrier transport though the disordered regions.40 This model is based on the assumption that the polymers which lack such a network ultimately result in a lower transistor performance. Another proposed mechanism is related to the polymer ordering. For CDT–BTZ polymers (2) it has been found that the film microstructure is independent of the molecular weight, but the crystallinity is significantly improved for higher Mn as evident from XRD data.33 As shown in Fig. 5a, the XRD diffraction peak for the highest Mn (35 kg mol−1, blue plots) is narrower than for polymers with lower Mn indicating higher crystallinity and a more pronounced polymer order for the first case. Additionally, the interlayer distance between backbones decreases from 2.78 nm for Mn = 11 kg mol−1 to 2.56 nm for Mn = 35 kg mol−1. A high order and tighter packing favor the charge carrier transport leading to a maximum value of 3.3 cm2 V−1 s−1 for Mn = 35 kg mol−1. An identical trend has been observed for 5 as shown in Fig. 5b. The XRD diffraction intensity is enhanced with increasing molecular weight suggesting an increased crystallinity of the thin films.44 This increase in crystallinity is in good agreement with the Mn dependent mobility (Fig. 4).
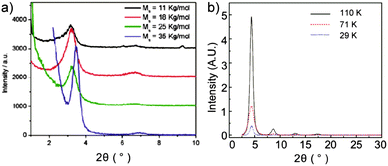 |
| | Fig. 5 X-ray diffraction of D–A polymer thin films of different molecular weights for (a) 2a33 (reprinted with permission from ref. 33, Copyright 2011, American Chemical Society) and (b) 543 (reprinted with permission from ref. 43, Copyright 2012, Nature Publishing Group). | |
However, some exceptions exist in which the molecular weight has no or negative impact on the charge carrier transport. In spite of improved molecular ordering with higher Mn, a quinacridone-based D–A polymer exhibited an Mn independent behavior of charge carrier transport in Mn ranges from 12 to 46 kg mol−1.45 Another unusual example is the n-type D–A polymer 1. It has been found that the low molecular weight derivative shows a superior electron mobility in comparison to the high molecular weight one, which is attributed to its higher crystallinity.46
4. Side chain engineering
As is mentioned in the previous sections, the device performance is typically strongly connected to the order of D–A polymers. Further aspects like supramolecular organization, local packing and microstructure formation are also important for the charge carrier transport and are determined mainly by the self-assembly propensity of the polymer and thus by intermolecular interactions. Due to these forces, the polymers can arrange into a three dimensional organization with long-range order which is necessary for an unhindered charge transport through the active layer. As a drawback, strong interactions between polymer chains can significantly reduce the solubility of these materials. To control the solubility and allow deposition of the polymers from solution, alkyl side chains are attached to the rigid backbone. Depending on the geometry of these substituents, the polymer interactions can be well controlled.47 Side chains also fulfill another important function. They take an essential role during the self-assembly of the polymer chains into distinct supramolecular structures. Typically, linear alkyl chains tend to form interchain interdigitation which promotes the formation of well-defined layered structures and increases the crystallinity as mainly proven for poly(alkylthiophenes).48 It is generally assumed that a highly crystalline layer structure favors the charge carrier transport. Interestingly, some D–A polymers revealed a contrary behavior.21
4.1. Linear vs. branched alkyl chains
To ensure sufficiently high solubility for device fabrication and good film formation of the rigid polymers branched side chains are usually chosen. Due to their higher steric hindrance these substituents lower the polymer interactions, can decrease the polymer planarity and rise the π-stacking distance within the layer structure.49 The π-stacking distance can therefore increase even by 0.5 Å significantly reducing the hopping rate of the charge carriers along the packing direction. Simulations have indicated that the electronic couplings decay exponentially with the stacking distance for cofacially π–π packed model systems and can vary even by a factor of 4 for an increase in packing distance of 0.5 Å.50 The relation between alkyl substituents and molecular organization has been discussed for CDT–BTZ polymers (2) for which the π-stacking distance increases from 3.5 Å to 4.0 Å by replacing the linear hexadecane alkyls (2a) by long branched decyl-tetradecyl ones (2d).51 In the case of 2d, the long branched decyl-tetradecyl chains surprisingly crystallize in the layer periphery inducing a pronounced microstructure in comparison with 2b with shorter branched ones. Due to the large π-stacking distance of 4.0 Å, the charge carrier mobility of 2b and 2d in FETs drops by several orders of magnitude in comparison to 2a. Interestingly, at the same time both polymers reveal an ambipolar behavior. This change in device operation in contrast to hole transporting 2a is related to a lateral shift of the backbones towards each other by 1–1.5 Å provoked by the high steric hindrance of the branched alkyl chains as indicated by solid-state NMR. Simulations have proven that this lateral shift equals the transfer integrals for holes and electrons on the same level from an initially hole dominated value of 2a.52 Besides the increase in stacking distance and backbone shift, the implementation of branched side chains can reduce the coherence lengths for the π-stacking and layer structure as a direct consequence of the lower backbone planarity.53 This in turn decreases the overall crystallinity that is crucial for the charge migration.
Another concept to tune the packing is based on “molecular docking” of isoindigo polymers 28 (Fig. 18) in which the spatial steric hindrance caused by branched alkyl chains is reduced by removal of these substituents just from the small units (Fig. 6).54 Following this approach, the smaller units dock into the cavity of the larger one accompanied by an improvement of the interchain π-stacking and carrier mobility. The packing is further enhanced by using centrosymmetric donor units yielding linear rather than curved backbone conformations (see further discussion on curvature in Section 5).
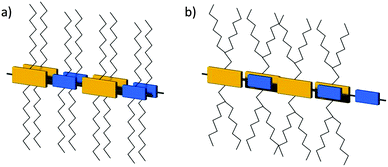 |
| | Fig. 6 Schematic illustration of polymers with (a) alkyl chains attached to donor and acceptor units, (b) molecular docking with substituents attached only to one donor or acceptor unit. | |
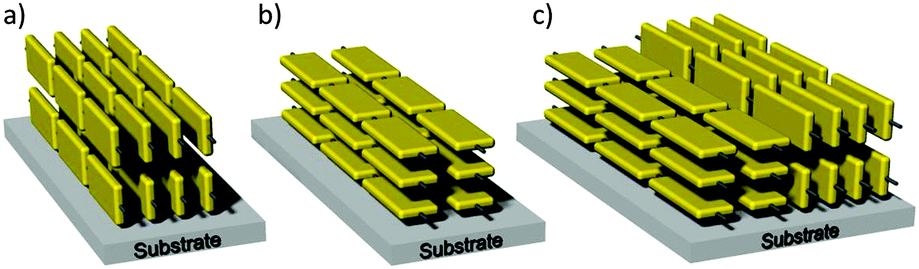
The side chain toolbox with linear, branched and hybrid substituents is also powerful to fine control the surface arrangement of the D–A polymers (Fig. 7). Besides the variation between edge-on and face-on55 the surface orientation can also include intermediate isotropic or bimodal states in active films (Fig. 7c). The backbone organization on the surface has tremendous impact on the charge carrier transport in the device. Charge transport in polymer films generally contains intrachain and interchain processes. The intrachain conduction is realized by π-electron delocalization along polymer backbones, providing high carrier mobility. This process is largely determined by the effective conjugation length of the polymer, which is limited by the torsional disorder along the backbone and the presence of chemical defects.56 The interchain transport is generally classified as a hopping mechanism. The most effective transport in transistors is achieved in the case of the edge-on organization with the π-stacking oriented parallel to the substrate and in the same plane of the charge transport. In a face-on organization the aromatic plane of the backbones lies flat on the surface so that the π-stacking is perpendicular to the substrate. This arrangement is beneficial in diode-like devices in a top and bottom electrode architecture as such heterojunction solar cells. The heterojunction morphology is typically formed from a mixture of a D–A polymer as donor system and a functionalized fullerene derivative as acceptor. The faster the holes and electrons are transported to the corresponding electrodes after exciton separation, the smaller the probability of recombination and finally the higher the solar cell efficiency. The conduction in diodes takes place out-of-plane to the surface which is in agreement with the π-stacking direction in a face-on organization.
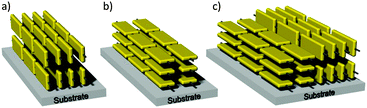 |
| | Fig. 7 Motifs of the polymer orientation on surfaces with (a) edge-on, (b) face-on, (c) bimodal arrangement. | |
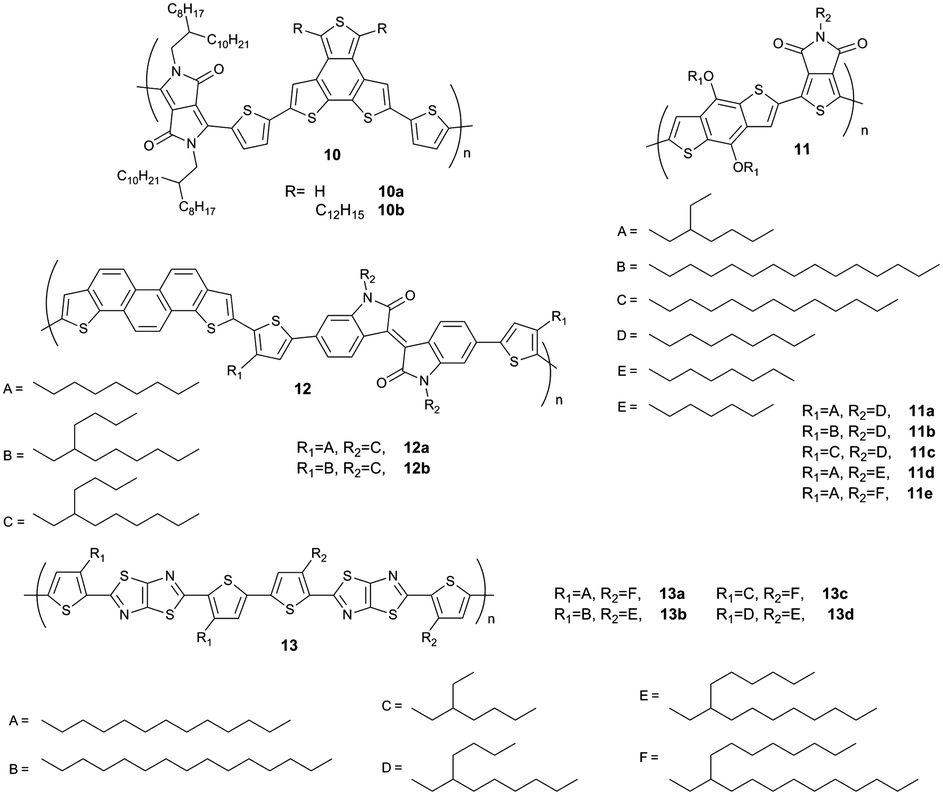
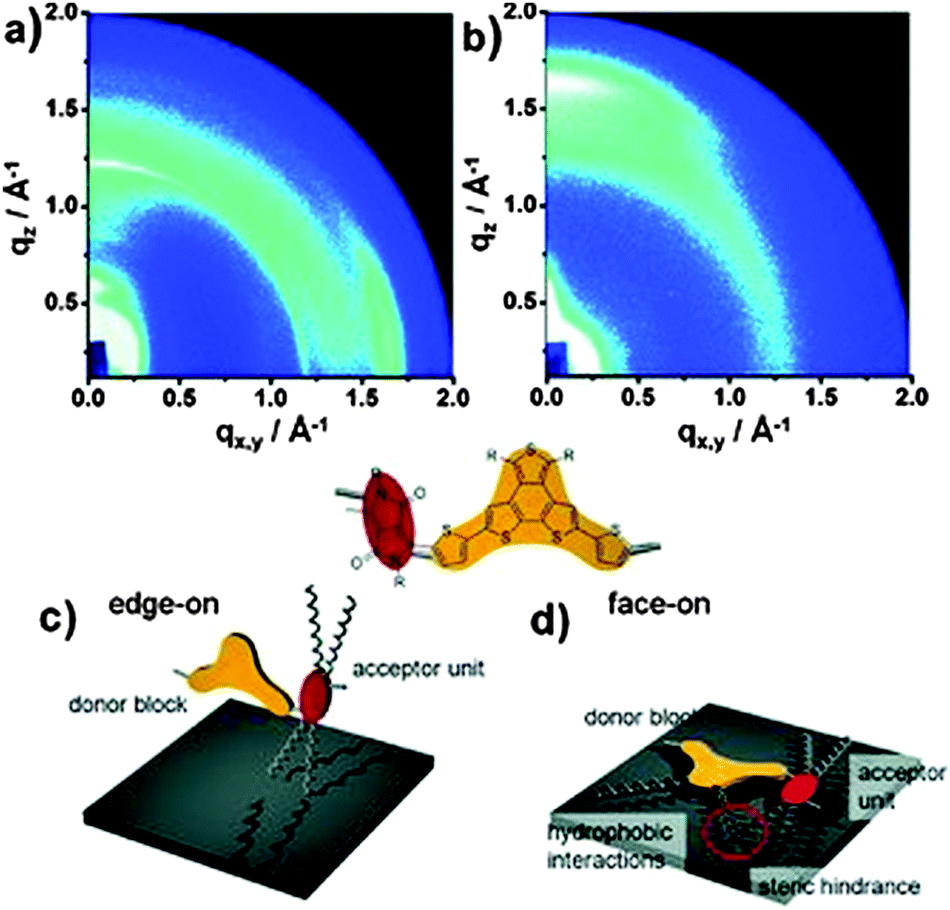
It has been shown for D–A polymers based on diketopyrrolopyrrole (10),57 thienopyrroledione (11)58 or phenanthrodithiophene–isoindigo (12)59 (Fig. 8) that bulky and sterically demanding side chains result in a face-on arrangement, while edge-on is triggered by linear ones in drop-cast and spin-coated films (Fig. 9). These changes in backbone orientation on the substrate are directly correlated to the device performance in transistors or solar cells. The mechanism in surface orientation is attributed to the polymer aggregation in solution before deposition. Strongly interacting polymer chains with linear substituents result in aggregates in solution which arrange edge-on on the surface. Upon poor aggregation single polymer chains present in the solution are flexible and capable to interact with the surface via van der Waals ordering in the face-on fashion. For polymer 13 the backbone orientation is altered through fine-tuning of the length difference between two alkyl substituents.60 For an edge-on orientation of 13, the length difference between the linear and branched side chain has to be large like in 13b and 13c. When the length difference is small, such as in 13a and 13d, the macromolecules tend to form a well-ordered face-on assembly. Bulk heterojunction solar cells reveal the highest short-circuit current density for films with primarily face-on orientation.
 |
| | Fig. 8 Chemical structures of D–A polymers with linear and branched alkyl substituents. | |
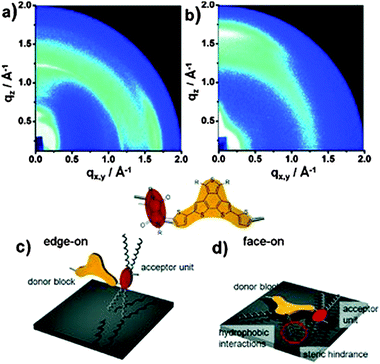 |
| | Fig. 9 GIWAXS patterns of (a) 10a and (b) 10b, schematic illustration for one repeating unit in a (c) edge-on for 10a and (d) face-on for 10b orientation towards the HMDS modified surface. The red circular areas indicate the steric hindrance between alkyl chains. Reprinted with permission from ref. 57, Copyright 2013, Wiley-VCH Verlag GmbH. | |
4.2. Role of bifurcation point
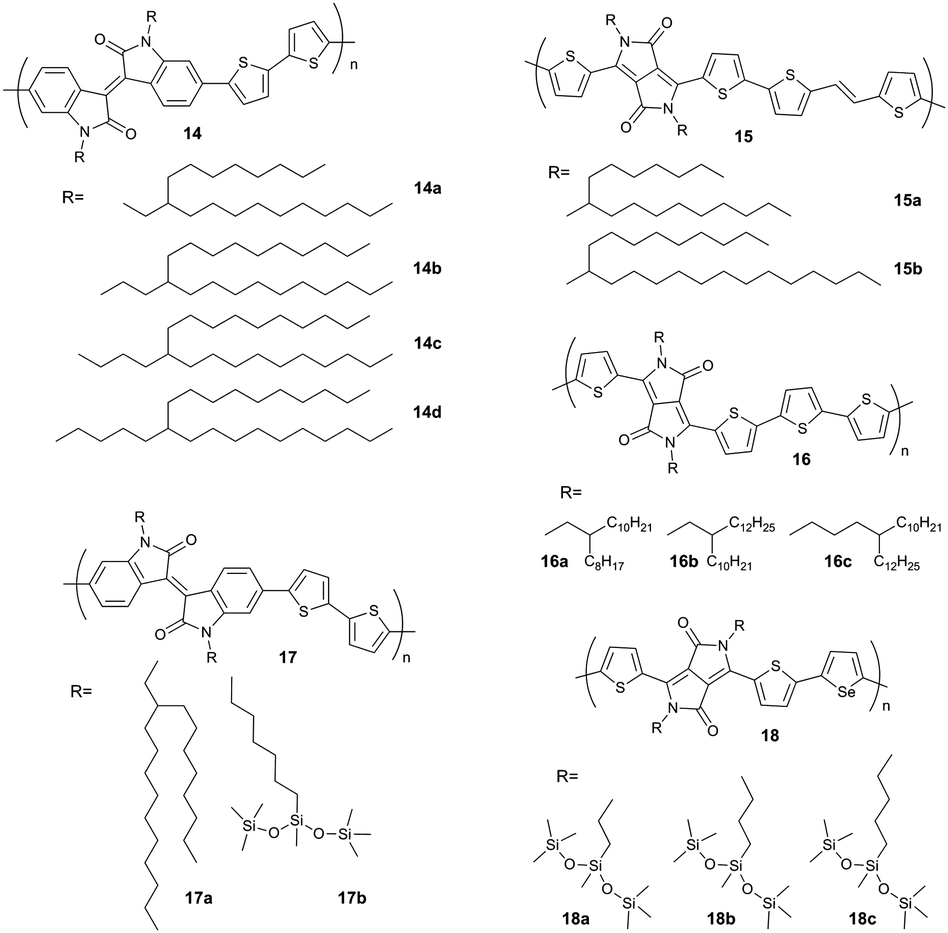
To overcome the steric impact of the branched alkyl substituents on the polymer packing, but to maintain the solubility, one strategy is to shift the branching point away from the backbone. Side chains with a branching point in proximity to the backbone hamper particularly the π-stacking interactions and lower the hopping rate of charge carriers. Systematic studies have shown that the spacer length between the branching point and the backbone plays an important role on polymer packing and transport of charge carriers.61,62 The reduced steric hindrance of attached alkyls allows closer π-stacking and lowers the polymer twisting leading to a more planar backbone.63 For instance, the π-stacking distance decreases from 3.75 Å for 14a to 3.57 Å for 14c and 14d, while at the same time the interlayer distance of edge-on oriented polymers increases with longer alkyl chains.
The same concept has been successfully applied for CDT-BTZ polymers (2). The 5-ethylnonyl side chains in 2c result in improved supramolecular organization in comparison to 2b with 2-ethylhexyls.64 Significant differences are also evident in the film microstructure. Polymer 2b possesses a lower solubility leading to the formation of 80 nm to 200 nm large aggregates, while a nanofiber network (diameter of 10 nm and length of about 100 nm) is obtained for 2c. The mobility for 2c is more than one order of magnitude higher than for 2b due to a smaller π-stacking distance and the fiber formation. The transport in 2b is disturbed by distinct grain boundaries, while nanofibers of 2c serve as good pathways for charge carriers allowing a 2D migration through the network.
A similar trend has been also observed for DPP polymers. A charge carrier mobility of 8.2 cm2 V−1 s−1 has been determined for a polymer with decyltetradecyl substituents (15a) which is two times larger than for the derivative with shorter octyldodecyl side chains (15b) (Fig. 10).65 This mobility increase is attributed also to stronger polymer interactions and a smaller π-stacking distance of 0.36 nm for 15a in comparison to 0.37 nm for 15b. Apart from the reduced π-stacking distance, the large size and high crystallinity of the domains improve the mobility, as well. The influence of branched alkyl chains and bifurcation point has been also investigated for other DPP-based polymers (16).66 Although the crystallinity of the DPP derivatives 16a and 16b (Fig. 10) is similar and the π-stacking distance for 16a is smaller, 16b shows a higher charge carrier mobility within the series of 16. The enhanced performance of 16b is assigned to its homogenous film and better interconnected network ensuring a more efficient charge transport between grains. Longer alkyl chains in 16c in comparison to 16b and 16a reduces the crystallinity, but increased the charge carrier mobility due to a shorter π-stacking distance. As in the other examples, the lowered π-stacking distance and increasing the backbone planarity are induced by shifting the bifurcation point.
 |
| | Fig. 10 Chemical structures of D–A polymers chosen to discuss the influence of the branching point and hybrid composition of the substituents. | |
4.3. Inorganic substituents
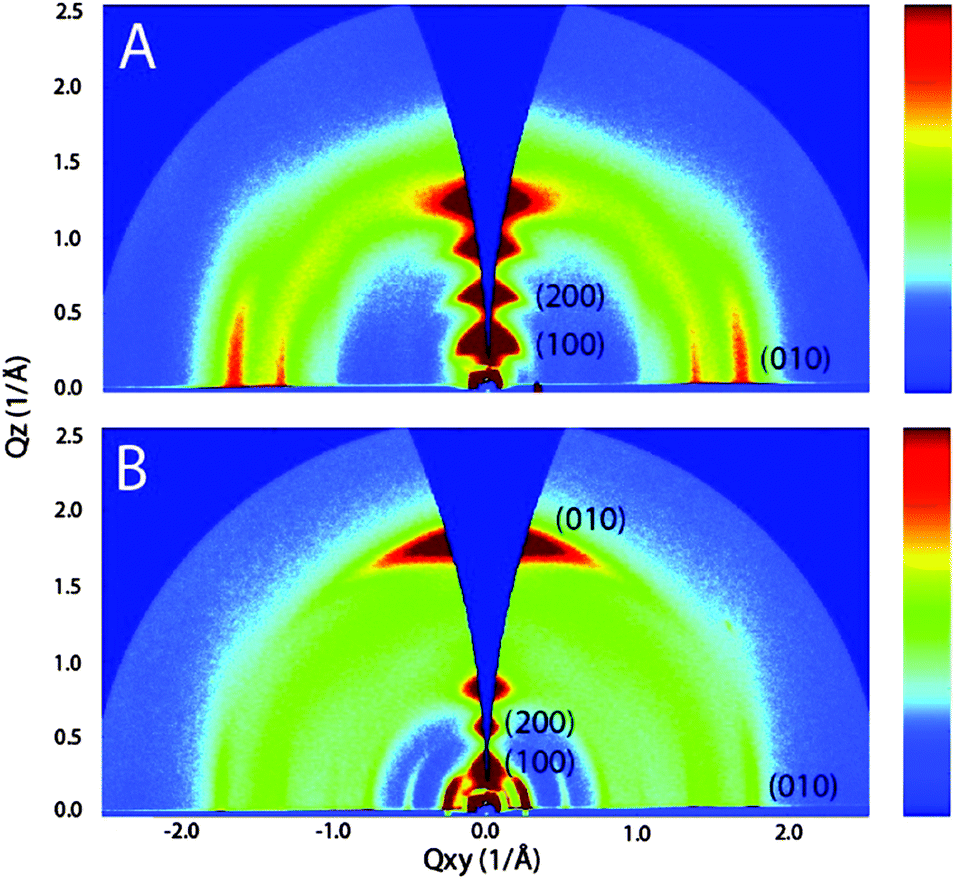
An identical correlation between branching point and packing has been found for D–A polymers siloxane-terminated substituents. The first approach has been reported for hybrid siloxane-terminated groups attached at an isoindigo-based polymer 17b ensuring a sufficient solubility for solution processing (Fig. 10).67 In comparison to a reference polymer 17a with branched alkyl side chains the π-stacking distance was reduced from 3.76 Å to 3.58 Å due to an enhanced backbone planarity leading to an increase of the hole mobility in transistors from 0.57 cm2 V−1 s−1 to 2.48 cm2 V−1 s−1. An additional factor responsible for the device improvement was a bimodal edge- and face-on arrangement of 17b on the surface allowing a 3D charge transport to by-pass structural defects through the active film (Fig. 11).
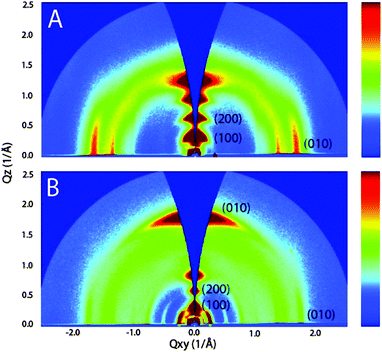 |
| | Fig. 11 GIWAXS patterns of 17a (A) and 17b (B). Polymer 17a displays lamellar packing common to many conjugated polymer films, with the π-stacking Bragg planes parallel to the substrate plane. In contrast, 17b crystallites contain two kinds of textures, where the π-stacking planes are both normal and parallel to the substrate.67 Reprinted with permission from ref. 67, Copyright 2011, American Chemical Society. | |
Similar to pure hydrocarbon substituents in 14, an important role of the spacer length has been found for the hybrid siloxane side chains on the organization and charge transport on transistors. Thereby, the ambipolar performance of DPP–selenophene polymers 18 is optimized by adjusting the branching position of siloxane substituents (Fig. 10). Polymers with reduced spacer length 18a and 18b exhibit enhanced charge transport relative to 20c due to a smaller lamellar spacing and a denser network of fibers in the film (Fig. 12), while retaining a close π-stacking distance.68 Solution-sheared films of 18b processed at elevated temperatures show unprecedentedly high hole and electron mobilities of 8.84 cm2 V−1 s−1 and 4.34 cm2 V−1 s−1, respectively.
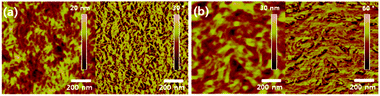 |
| | Fig. 12 AFM height (left) and phase (right) images of solution-processed drop-cast films of (a) 18a and (b) 18b.68 Reprinted with permission from ref. 68, Copyright 2013, American Chemical Society. | |
4.4. Odd–even effect of substituents
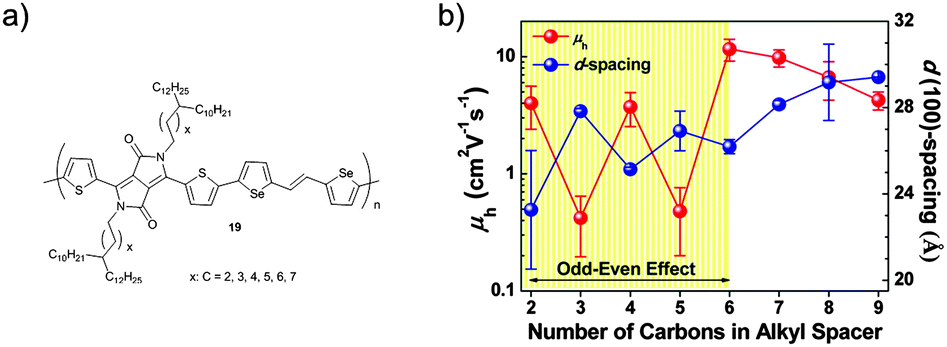
Not only the spacer length, but also its odd–even carbon number has impact on the self-organization and electrical properties of D–A polymers (Fig. 13). Polymer derivatives 19 with branched alkyl groups containing linear spacers from C2 to C9 show such an odd–even effect.69 Alkyl spacers with even numbers of carbon atoms (C2, C4, and C6) exhibit shorter lattice spacing than spacers with odd carbon numbers (C3 and C5) resulting in one order of magnitude higher charge mobilities. It is assumed that various torsion angles with respect to the alkyl chain branching position affect the polymer packing resulting in longer/shorter d-spacing upon odd/even numbered spacers. The denser packing of 19 with even numbered spacers (C2, C4, and C6) implies that their alkyl side chains might undergo relatively strong interdiffusion reducing the intermolecular spacing of the polymer backbones. As the side chain length increases the odd–even effect fades for lengths larger than C7. The longer alkyl spacer may shield the odd–even effect for the polymer packing due to their unpredictable interaction with neighboring segments.
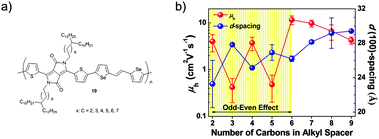 |
| | Fig. 13 (a) Chemical structure of 19 revealing an odd–even effect of the alkyl spacer, (b) average mobility and d(100)-spacing values of 19 as a function of carbon number in the spacer.69 Reprinted with permission from ref. 69, Copyright 2015, American Chemical Society. | |
4.5. Substituent position
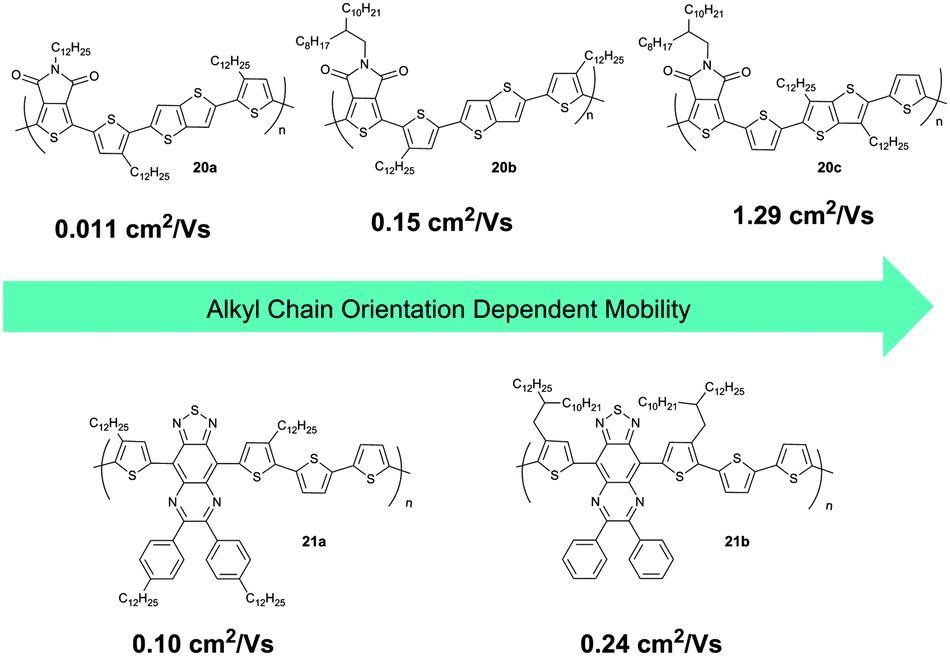
Besides the geometry of the side chains, their substitution position at the backbone plays also an important role on the supramolecular organization of D–A polymers. For instance, the variation in alkyl chain orientation at the rigid backbones significantly influences the packing and charge carrier transport of three thienopyrroledione-based polymers 20 with a similar alkyl chain density, but differing in the side chain position (Fig. 14).70 Polymer 20b possesses a large space between clusters of alkyl chains and facilitates in this way interdigitation, formation of layer structures and large intermolecular overlap. In contrast, a small space between alkyl clusters and non-uniform chain orientation in 20a and 20c hinders interdigitation and intermolecular overlap. For this reason, the mobility of 20c is more than two orders of magnitude higher in comparison to 20a and 20b. The geometry and localization of linear and branched substituents have been also investigated for thiadiazoloquinoxaline-based polymers 21. A pair of 2-decyltetradecyl chains in 21b used to replace the linear side chains in 21a does not significantly change the planarity of the polymers, but improves the molecular weight and solubility. In this way, the films are more homogenous and show higher crystallinity in the π-stacking direction of the polymer. Therefore, the charge carrier mobility of 21b increases up to 0.24 cm2 V−1 s−1 with respect to 21a (Fig. 14).
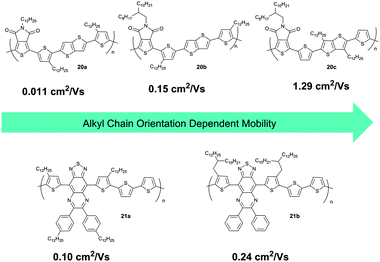 |
| | Fig. 14 Relation between charge carrier mobility and alkyl chain position in selected D–A polymers. | |
4.6. Thermally controlled substituents
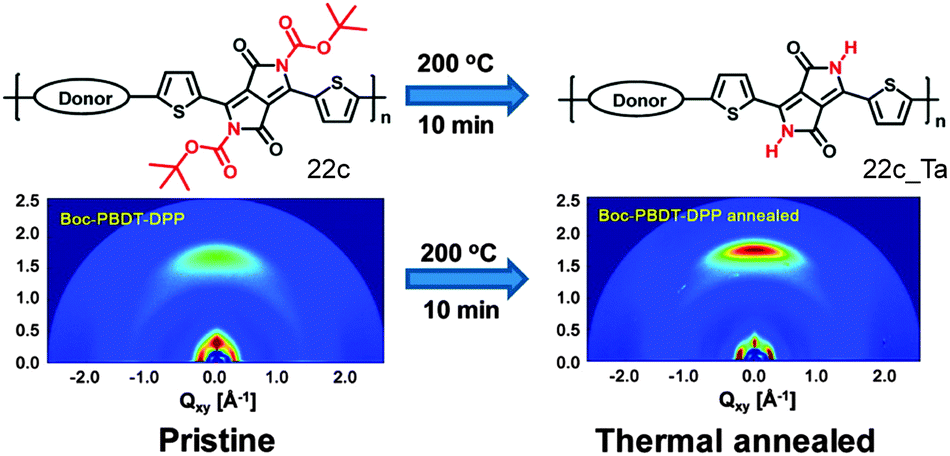
One recent strategy has used thermally removable substituents to turn the polymer film insoluble for subsequent film deposition and to reduce the portion of isolating alkyl chains in the active layer. The main requirement is that the electronic nature of the polymer backbone remains unperturbed by the bond scission. The cleavage mechanism of the substituents has been well described for polythiophenes as model systems71 and the approach has been recently transferred to a series of narrow band gap D–A polymers comprising tert-butoxycarbonyl (t-Boc) substituted indigo (22a), isoindigo (22b) or diketopyrrolopyrrole (22c) as the acceptor and benzodithiophene as donor moiety (Fig. 15).72 Thermal treatment of 22c films at 200 °C leads to the cleavage of the t-Boc side groups along with the formation of a hydrogen-bonding network and enhanced backbone planarity due to a reduction of the steric hindrance. A higher planarity facilitates intermolecular interactions between fused ring moieties and the thiophene segments. For this reason, the interlayer and π-planar distances decrease after thermal reduction being beneficial for the charge carrier transport in transistors (Fig. 16).73
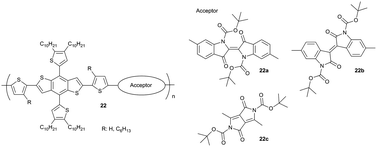 |
| | Fig. 15 Chemical structures of D–A polymer with thermally removable substituents. | |
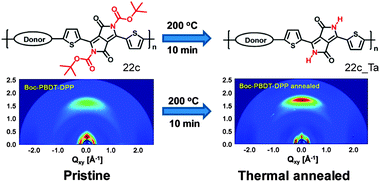 |
| | Fig. 16 Schematic illustration of the thermal reduction of the substituents in 22c and GIWAXS patterns obtained before and after substituent reduction. Reprinted with permission from ref. 72, Copyright 2015, American Chemical Society. | |
5. Role of backbone curvature
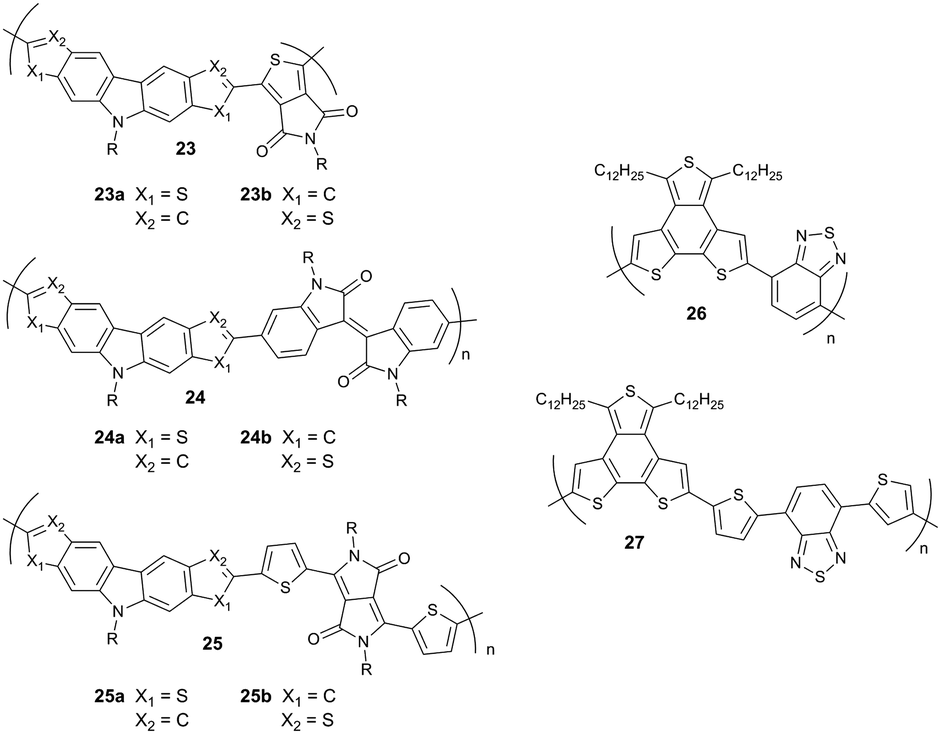
The backbone curvature can significantly impact the supramolecular organization of D–A polymers. For instance, polythiophene derivatives with increased backbone curvature result during synthesis generally in low molecular weight. The effect of molecular weight reduction is attributed to difficulties during polymerization by reducing the accessibility of the reactive chain ends for further reactions. The flexibility of polymers is strongly connected to their solubility which is also affected by the backbone curvature. On the one hand, the polymer solubility is increased with higher backbone curvature resulting in reduced aggregation of the rod-like polymers. On the other hand, a too high curvature hinders lamellar packing due to higher entropy. In the solid-state, an increased interlayer distance is typically observed for more curved polymers since side chains on the bent backbones cannot interdigitate as effectively as in the case of linear polymers.74 The influence of interdigitation on the organization and charge carrier transport has been already discussed in a previous section.74 The role of different bonding geometries of dithieno-carbazole-based D–A polymers on the backbone conformation has been studied for a series of derivatives with various donor and acceptor units (23–25). Polymers 23a, 24a and 25a show a strong backbone curvature and in consequence, form amorphous films associated with relatively low charge carrier mobilities in the range of 10−3 cm2 V−1 s−1. Polymers 23b and 25b possess pseudo-straight-shaped backbones and yield ordered films with an edge-on orientation. Accordingly, 23b and 25b exhibit the highest mobilities within the series of 0.31 and 1.36 cm2 V−1 s−1, respectively. A lower mobility of 24b in comparison to 23b and 25b was assigned to the stiff backbone which resulted in poor self-assembly and a low-ordered film (Fig. 17).75
 |
| | Fig. 17 Chemical structures of D–A polymers with different backbone curvatures influencing the organization. | |
Changes in the organization of D–A polymers related to differently shaped curvatures induced by additional thiophene units in the backbone have been reported for 26 and 27.57 Polymer 26 reveals a similar organization as observed for discotic columnar systems which is related to the disk-like shape of the donor unit packed on top of each other to form a columnar stack.76 Additionally, 26 assembles in a face-on fashion on the substrate. In contrast, 27 organizes in edge-on oriented layers leading to a higher charge carrier mobility in transistors.57 The strong backbone curvature in high mobility D–A polymers can be avoided by the right selection of the donor and acceptor units.75 The role of the chemical structure of the donor unit on the backbone curvature has been described for centro- and axisymmetric units (Fig. 18).54 Among these two systems, polymer 28a with a centrosymmetric donor exhibits a low backbone curvature and therefore high crystallinity, improved lamellar packing and good interchain π-stacking leading to high a mobility around 1 cm2 V−1 s−1. Thereby, the packing is further favored by the “molecular docking” process described in Section 4.1 when the small unit docks into the cavity of the larger acceptor core (Fig. 6). Additional methyl groups at the donor lower the order of 28b so that the mobility drops to 0.11 cm2 V−1 s−1 due to poor π-stacking interactions between polymer chains which nevertheless retain a linear backbone and a good lamellar packing. This value further decreases to 0.061 cm2 V−1 s−1 for 28c and 28d containing axisymmetric donors due to their higher backbone curvature which inhibits docking and reduces lamellar order (Fig. 19).
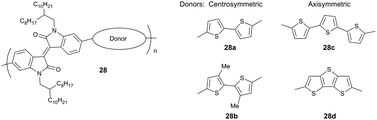 |
| | Fig. 18 Chemical structures of isoindigo-based polymers with centrosymmetric and axisymmetric donor units. | |
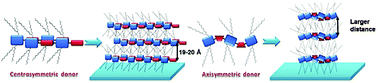 |
| | Fig. 19 Illustration of the film packings for 28 with centrosymmetric and axisymmetric donors. Reprinted with permission from ref. 54, Copyright 2012, American Chemical Society. | |
6. Conclusions
Conjugated D–A polymers provide a high degree of design freedom including the bulkiness of the substituents as well as structure factors like backbone curvature and molecular weight. Alkyl substituents ensure solubility which is required for the solution processing and microstructure formation in thin films. Additionally, substituents play an important role for the packing of the polymers and the backbone planarity. Higher steric demand of side chains which can arise from their geometry or from the substitution position at the backbone can significantly increase the π-stacking distance between conjugated backbones leading to a decrease in the hopping rate along the stacking direction. It is necessary to find the right balance between steric demand of the substituents, polymer packing and backbone twisting. Too bulky substituents provoke a backbone twist resulting in reduced order and lowered charge carrier migration along the conjugated polymer chain. Apparently, substituents seem to influence also the polymer surface arrangement in thin films. Depending on the type and position of the alkyls the organization can be switched between face-on and edge-on. The control over the surface arrangement is important for the implementation of polymers in electronic devices of different geometry.
Comprehensive understanding of the influence of chemical design of D–A polymers on their packing and assembly has been gained and is essential for the development of future high performance organic semiconductors. Identification of key conditions for a defect-free structure formation during solution processing allows a distinct control over the arrangement and orientation of D–A polymers on surfaces opening the door to novel large-scale roll-to-roll technologies.
Acknowledgements
This work was supported by the National Science Centre, Poland, through the grant UMO-2015/18/E/ST3/00322. We are grateful to Klaus Müllen and Wojciech Zajaczkowski for stimulating discussions.
Notes and references
- Y. Ohmori, Laser Photonics Rev., 2010, 4, 300–310 CrossRef CAS.
- B. Geffroy, P. le Roy and C. Prat, Polym. Int., 2006, 55, 572–582 CrossRef CAS.
- R. A. Street, Adv. Mater., 2009, 21, 2007–2022 CrossRef CAS.
- H. Sirringhaus, Adv. Mater., 2014, 26, 1319–1335 CrossRef CAS PubMed.
- C. J. Brabec, N. S. Sariciftci and J. C. Hummelen, Adv. Funct. Mater., 2001, 11, 15–26 CrossRef CAS.
- S. Allard, M. Forster, B. Souharce, H. Thiem and U. Scherf, Angew. Chem., Int. Ed., 2008, 47, 4070–4098 CrossRef CAS PubMed.
- Z. Bao, Adv. Mater., 2000, 12, 227–230 CrossRef CAS.
- T. Sekitani, U. Zschieschang, H. Klauk and T. Someya, Nat. Mater., 2010, 9, 1015–1022 CrossRef CAS PubMed.
- D. J. Gundlach, Nat. Mater., 2007, 6, 173–174 CrossRef CAS PubMed.
- A. N. Sokolov, M. E. Roberts and Z. Bao, Mater. Today, 2009, 12, 12–20 CrossRef.
- S. Holliday, J. E. Donaghey and I. McCulloch, Chem. Mater., 2014, 26, 647–663 CrossRef CAS.
- P. M. Beaujuge and J. M. J. Fréchet, J. Am. Chem. Soc., 2011, 133, 20009–20029 CrossRef CAS PubMed.
- X. Guo, R. P. Ortiz, Y. Zheng, Y. Hu, Y.-Y. Noh, K.-J. Baeg, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2011, 133, 1405–1418 CrossRef CAS PubMed.
- A. Facchetti, Mater. Today, 2007, 10, 28–37 CrossRef CAS.
- G. M. Whitesides and M. Boncheva, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4769–4774 CrossRef CAS PubMed.
- A. Mishra, C.-Q. Ma and P. Bäuerle, Chem. Rev., 2009, 109, 1141–1276 CrossRef CAS PubMed.
- T. Lei, Y. Cao, Y. Fan, C.-J. Liu, S.-C. Yuan and J. Pei, J. Am. Chem. Soc., 2011, 133, 6099–6101 CrossRef CAS PubMed.
- I. McCulloch, C. Bailey, M. Giles, M. Heeney, I. Love, M. Shkunov, D. Sparrowe and S. Tierney, Chem. Mater., 2005, 17, 1381–1385 CrossRef CAS.
- A. Facchetti, Chem. Mater., 2011, 23, 733–758 CrossRef CAS.
- A. Salleo, Mater. Today, 2007, 10, 38–45 CrossRef CAS.
- R. Noriega, J. Rivnay, K. Vandewal, F. P. V. Koch, N. Stingelin, P. Smith, M. F. Toney and A. Salleo, Nat. Mater., 2013, 12, 1038–1044 CrossRef CAS PubMed.
- N.-K. Kim, S.-Y. Jang, G. Pace, M. Caironi, W.-T. Park, D. Khim, J. Kim, D.-Y. Kim and Y.-Y. Noh, Chem. Mater., 2015, 27, 8345–8353 CrossRef CAS.
- S. R. Forrest, Nature, 2004, 428, 911–918 CrossRef CAS PubMed.
- H. N. Tsao and K. Müllen, Chem. Soc. Rev., 2010, 39, 2372–2386 RSC.
- L. Biniek, B. C. Schroeder, C. B. Nielsen and I. McCulloch, J. Mater. Chem., 2012, 22, 14803–14813 RSC.
- J. Peet, J. Y. Kim, N. E. Coates, W. L. Ma, D. Moses, A. J. Heeger and G. C. Bazan, Nat. Mater., 2007, 6, 497–500 CrossRef CAS PubMed.
- J. Y. Kim, K. Lee, N. E. Coates, D. Moses, T.-Q. Nguyen, M. Dante and A. J. Heeger, Science, 2007, 317, 222–225 CrossRef CAS PubMed.
- J. S. Lee, S. K. Son, S. Song, H. Kim, D. R. Lee, K. Kim, M. J. Ko, D. H. Choi, B. Kim and J. H. Cho, Chem. Mater., 2012, 24, 1316–1323 CrossRef CAS.
- C. An, M. Li, T. Marszalek, D. Li, R. Berger, W. Pisula and M. Baumgarten, Chem. Mater., 2014, 26, 5923–5929 CrossRef CAS.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed.
- I. Kang, H.-J. Yun, D. S. Chung, S.-K. Kwon and Y.-H. Kim, J. Am. Chem. Soc., 2013, 135, 14896–14899 CrossRef CAS PubMed.
- B. Sun, W. Hong, Z. Yan, H. Aziz and Y. Li, Adv. Mater., 2014, 26, 2636–2642 CrossRef CAS PubMed.
- H. N. Tsao, D. M. Cho, I. Park, M. R. Hansen, A. Mavrinskiy, D. Y. Yoon, R. Graf, W. Pisula, H. W. Spiess and K. Müllen, J. Am. Chem. Soc., 2011, 133, 2605–2612 CrossRef CAS PubMed.
- M. Brinkmann, E. Gonthier, S. Bogen, K. Tremel, S. Ludwigs, M. Hufnagel and M. Sommer, ACS Nano, 2012, 6, 10319–10326 CrossRef CAS PubMed.
- C. L. Donley, J. Zaumseil, J. W. Andreasen, M. M. Nielsen, H. Sirringhaus, R. H. Friend and J.-S. Kim, J. Am. Chem. Soc., 2005, 127, 12890–12899 CrossRef CAS PubMed.
- W. Zhang, J. Smith, S. E. Watkins, R. Gysel, M. McGehee, A. Salleo, J. Kirkpatrick, S. Ashraf, T. Anthopoulos, M. Heeney and I. McCulloch, J. Am. Chem. Soc., 2010, 132, 11437–11439 CrossRef CAS PubMed.
- T.-Y. Chu, J. Lu, S. Beaupré, Y. Zhang, J.-R. Pouliot, J. Zhou, A. Najari, M. Leclerc and Y. Tao, Adv. Funct. Mater., 2012, 22, 2345–2351 CrossRef CAS.
- X. Zhang, H. Bronstein, A. J. Kronemeijer, J. Smith, Y. Kim, R. J. Kline, L. J. Richter, T. D. Anthopoulos, H. Sirringhaus, K. Song, M. Heeney, W. Zhang, I. McCulloch and D. M. DeLongchamp, Nat. Commun., 2013, 4, 2238 Search PubMed.
- J. J. Intemann, K. Yao, H.-L. Yip, Y.-X. Xu, Y.-X. Li, P.-W. Liang, F.-Z. Ding, X. Li and A. K. Y. Jen, Chem. Mater., 2013, 25, 3188–3195 CrossRef CAS.
- B. Fu, J. Baltazar, Z. Hu, A.-T. Chien, S. Kumar, C. L. Henderson, D. M. Collard and E. Reichmanis, Chem. Mater., 2012, 24, 4123–4133 CrossRef CAS.
- H. N. Tsao, D. M. Cho, I. Park, M. R. Hansen, A. Mavrinskiy, Y. Yoon do, R. Graf, W. Pisula, H. W. Spiess and K. Müllen, J. Am. Chem. Soc., 2011, 133, 2605–2612 CrossRef CAS PubMed.
- Y. Li, P. Sonar, S. P. Singh, M. S. Soh, M. van Meurs and J. Tan, J. Am. Chem. Soc., 2011, 133, 2198–2204 CrossRef CAS PubMed.
- J. Li, Y. Zhao, H. S. Tan, Y. Guo, C.-A. Di, G. Yu, Y. Liu, M. Lin, S. H. Lim, Y. Zhou, H. Su and B. S. Ong, Sci. Rep., 2012, 2, 754 Search PubMed.
- J. Li, Y. Zhao, H. S. Tan, Y. Guo, C. A. Di, G. Yu, Y. Liu, M. Lin, S. H. Lim, Y. Zhou, H. Su and B. S. Ong, Sci. Rep., 2012, 2, 754 Search PubMed.
- I. Osaka, M. Akita, T. Koganezawa and K. Takimiya, Chem. Mater., 2012, 24, 1235–1243 CrossRef CAS.
- Y. Karpov, W. Zhao, I. Raguzin, T. Beryozkina, V. Bakulev, M. Al-Hussein, L. Häußler, M. Stamm, B. Voit, A. Facchetti, R. Tkachov and A. Kiriy, ACS Appl. Mater. Interfaces, 2015, 7, 12478–12487 CAS.
- J. Mei and Z. Bao, Chem. Mater., 2014, 26, 604–615 CrossRef CAS.
- D. M. DeLongchamp, R. J. Kline, E. K. Lin, D. A. Fischer, L. J. Richter, L. A. Lucas, M. Heeney, I. McCulloch and J. E. Northrup, Adv. Mater., 2007, 19, 833–837 CrossRef CAS.
- I. Osaka, R. Zhang, G. Sauvé, D.-M. Smilgies, T. Kowalewski and R. D. McCullough, J. Am. Chem. Soc., 2009, 131, 2521–2529 CrossRef CAS PubMed.
- V. Coropceanu, J. Cornil, D. A. da Silva Filho, Y. Olivier, R. Silbey and J.-L. Brédas, Chem. Rev., 2007, 107, 926–952 CrossRef CAS PubMed.
- W. Pisula, H. Tsao, D. Dudenko, D. Cho, S. Puniredd, Y. Zhao, A. Mavrinskiy, J. Shu, M. Hansen, M. Baumgarten and K. Müllen, Polymers, 2013, 5, 833 CrossRef.
- Y. Olivier, D. Niedzialek, V. Lemaur, W. Pisula, K. Müllen, U. Koldemir, J. R. Reynolds, R. Lazzaroni, J. Cornil and D. Beljonne, Adv. Mater., 2014, 26, 2119–2136 CrossRef CAS PubMed.
- A. T. Yiu, P. M. Beaujuge, O. P. Lee, C. H. Woo, M. F. Toney and J. M. J. Fréchet, J. Am. Chem. Soc., 2012, 134, 2180–2185 CrossRef CAS PubMed.
- T. Lei, Y. Cao, X. Zhou, Y. Peng, J. Bian and J. Pei, Chem. Mater., 2012, 24, 1762–1770 CrossRef CAS.
- J. Guo, Y. Liang, J. Szarko, B. Lee, H. J. Son, B. S. Rolczynski, L. Yu and L. X. Chen, J. Phys. Chem. B, 2010, 114, 742–748 CrossRef CAS PubMed.
- F. C. Grozema, P. T. van Duijnen, Y. A. Berlin, M. A. Ratner and L. D. A. Siebbeles, J. Phys. Chem. B, 2002, 106, 7791–7795 CrossRef CAS.
- X. Guo, S. R. Puniredd, M. Baumgarten, W. Pisula and K. Müllen, Adv. Mater., 2013, 25, 5467–5472 CrossRef CAS PubMed.
- C. Cabanetos, A. El Labban, J. A. Bartelt, J. D. Douglas, W. R. Mateker, J. M. Frechet, M. D. McGehee and P. M. Beaujuge, J. Am. Chem. Soc., 2013, 135, 4656–4659 CrossRef CAS PubMed.
- S. Nishinaga, H. Mori and Y. Nishihara, Macromolecules, 2015, 48, 2875–2885 CrossRef CAS.
- I. Osaka, M. Saito, T. Koganezawa and K. Takimiya, Adv. Mater., 2014, 26, 331–338 CrossRef CAS PubMed.
- J. Lee, A. R. Han, J. Kim, Y. Kim, J. H. Oh and C. Yang, J. Am. Chem. Soc., 2012, 134, 20713–20721 CrossRef CAS PubMed.
- J. Lee, A. R. Han, H. Yu, T. J. Shin, C. Yang and J. H. Oh, J. Am. Chem. Soc., 2013, 135, 9540–9547 CrossRef CAS PubMed.
- T. Lei, J. H. Dou and J. Pei, Adv. Mater., 2012, 24, 6457–6461 CrossRef CAS PubMed.
- J. Lee, T. Marszalek, K. C. Lee, J. Kim, W. Pisula and C. Yang, Macromol. Chem. Phys., 2015, 216, 1244–1250 CrossRef CAS.
- H. Chen, Y. Guo, G. Yu, Y. Zhao, J. Zhang, D. Gao, H. Liu and Y. Liu, Adv. Mater., 2012, 24, 4618–4622 CrossRef CAS PubMed.
- S. Chen, B. Sun, W. Hong, H. Aziz, Y. Meng and Y. Li, J. Mater. Chem. C, 2014, 2, 2183 RSC.
- J. Mei, H. Kim do, A. L. Ayzner, M. F. Toney and Z. Bao, J. Am. Chem. Soc., 2011, 133, 20130–20133 CrossRef CAS PubMed.
- J. Lee, A. R. Han, H. Yu, T. J. Shin, C. Yang and J. H. Oh, J. Am. Chem. Soc., 2013, 135, 9540–9547 CrossRef CAS PubMed.
- J. Y. Back, H. Yu, I. Song, I. Kang, H. Ahn, T. J. Shin, S.-K. Kwon, J. H. Oh and Y.-H. Kim, Chem. Mater., 2015, 27, 1732–1739 CrossRef CAS.
- Q. Wu, M. Wang, X. Qiao, Y. Xiong, Y. Huang, X. Gao and H. Li, Macromolecules, 2013, 46, 3887–3894 CrossRef CAS.
- M. Kuhn, J. Ludwig, T. Marszalek, T. Adermann, W. Pisula, K. Müllen, A. Colsmann and M. Hamburger, Chem. Mater., 2015, 27, 2678–2686 CrossRef CAS.
- C. Liu, S. Dong, P. Cai, P. Liu, S. Liu, J. Chen, F. Liu, L. Ying, T. P. Russell, F. Huang and Y. Cao, ACS Appl. Mater. Interfaces, 2015, 7, 9038–9051 CAS.
- J. Lee, A. R. Han, J. Hong, J. H. Seo, J. H. Oh and C. Yang, Adv. Funct. Mater., 2012, 22, 4128–4138 CrossRef CAS.
- R. Rieger, D. Beckmann, A. Mavrinskiy, M. Kastler and K. Müllen, Chem. Mater., 2010, 22, 5314–5318 CrossRef CAS.
- Y. Deng, Y. Chen, X. Zhang, H. Tian, C. Bao, D. Yan, Y. Geng and F. Wang, Macromolecules, 2012, 45, 8621–8627 CrossRef CAS.
- W. Pisula, X. A. Feng and K. Müllen, Adv. Mater., 2010, 22, 3634–3649 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a