
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanothermally induced conformational switch of a porphyrin dimer in a polymer film†
Hung
Doan
a,
Sangram L.
Raut
a,
David
Yale
a,
Milan
Balaz
*b,
Sergei V.
Dzyuba
*c and
Zygmunt
Gryczynski
*a
aDepartment of Physics and Astronomy, Texas Christian University, Fort Worth, TX 76129, USA. E-mail: z.gryczynski@tcu.edu
bUnderwood International College, Integrated Science & Engineering Division, Yonsei University, Seoul, 03722, Republic of Korea. E-mail: mbalaz@yonsei.ac.kr
cDepartment of Chemistry and Biochemistry, Texas Christian University, Fort Worth, TX 76129, USA. E-mail: s.dzyuba@tcu.edu
First published on 7th June 2016
Abstract
Stretching a polymer film induces a conformational change (from the twisted to planar state) in the embedded porphyrin dimer, as evidenced by steady-state and time-resolved emission spectra.
A number of physicochemical properties, including reactivity and optical characteristics, of small molecules depend on their conformational preference. The external modulation of chemical and physical processes may offer extensive applications in a wide range of fields1 and could be an effective approach to induce particular conformations and thus properties in organic materials. Among conventional stimuli, such as heat, light, or electrical potential, the use of mechanical force constitutes a potentially useful, yet currently unexplored approach to control the conformational preference of organic molecules.2 On the other hand, mechanochemistry, i.e., the physical and chemical transformations of materials induced by mechanical forces such as stretching, grinding, or pressure, has become a burgeoning field in recent years.3
Typically, mechanofluorochromic behavior is achieved by modifying the physical state or chemical structure of a molecule under mechanical stress, where mechanophores are covalently bonded to a polymer matrix, and thus experience the impact of mechanical force due to changes imposed in the matrix.4 A complementary approach stems from structural changes in the physical supramolecular arrangement of molecules.5 To date, various compounds have been reported to show mechanical force-induced emission changes in the solid state using the abovementioned approaches.6 In addition, fluorophores in which a multicolor switch could be induced by mechanical force via the synergistic effect of intramolecular conformational twisting and intermolecular molecular packing modes were recently reported.7 Notably, the photophysical properties of 9,10-bis(phenylethynyl)anthracene and several poly(phenyleneethynylene)s were investigated in regard to the aggregation-induced coplanarization of the twisted species in solution and thin films.8 However, the conformational changes between twisted and planar forms were not attributed to the stretching of the films. Thus, the use of mechanical force to induce and stabilize a particular conformation, and thereby modulate the photophysical properties of a single small molecule (i.e., not the supramolecular assemblies of small molecules, polymers, oligomers), has yet to be established.
A conjugated porphyrin dimer (PD, Fig. 1) has garnered considerable attention as a viscometer and sensitizer in photodynamic therapy.9 Rotation around the diyne moiety results in a collection of conformations, owing to the relative orientation of the porphyrin units, with the twisted (θ = 90°) and planar (θ = 0°) conformations as the extremes. Notably, due to the triethylene glycol moieties on the porphyrin core, and the preferred non-planar orientation of the phenylene groups, the self-assembly/aggregation of PD is unlikely, and therefore the spectral properties are related to the particular state/conformation of an individual molecule, rather than to those of the aggregates.9
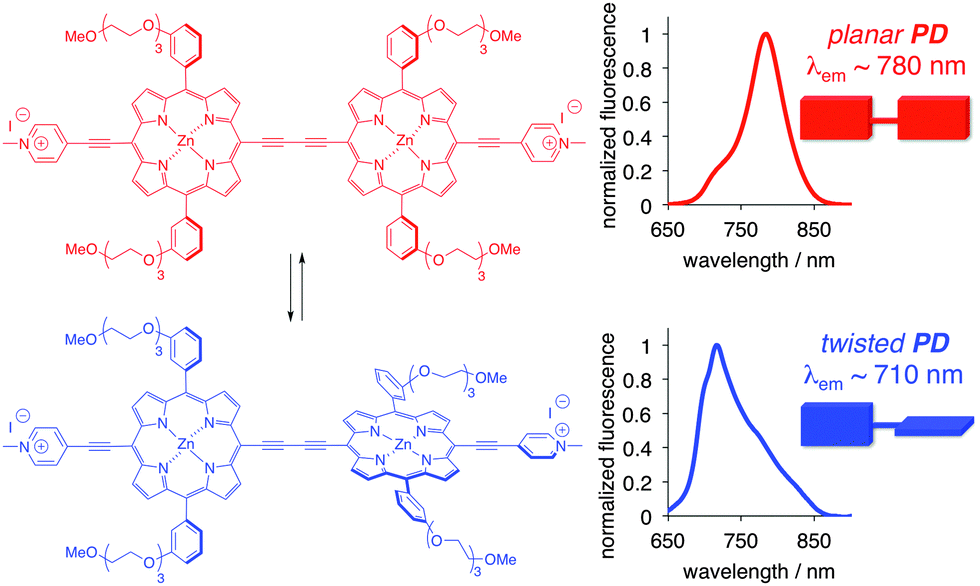 | ||
| Fig. 1 Structure and emission spectra of conformational extremes (planar and twisted) of PD. | ||
The twisted and planar conformations of PD exhibit distinct emission spectra, with emission maxima appearing at around 710 and 780 nm, respectively. The distinct photophysical properties of the two conformers allow for the facile assessment of PD's conformation in a variety of different media including organic solvents, ionic liquids, micellar, and cellular environments.9b,10 In all aforementioned examples, the conformation of PD was not locked, and dynamic equilibrium was established between the planar and twisted conformations depending on the nature of the media. Fluorescence spectroscopy was found to be particularly useful in accessing the conformer ratio as a function of the media's viscosity. Upon excitation, the excited twisted conformation emits and/or converts into a lower energy excited planar conformer in low viscosity media, whereas in high viscosity media, this process is not allowed, thus leading to the ground state of the twisted conformer (Fig. 2A).11
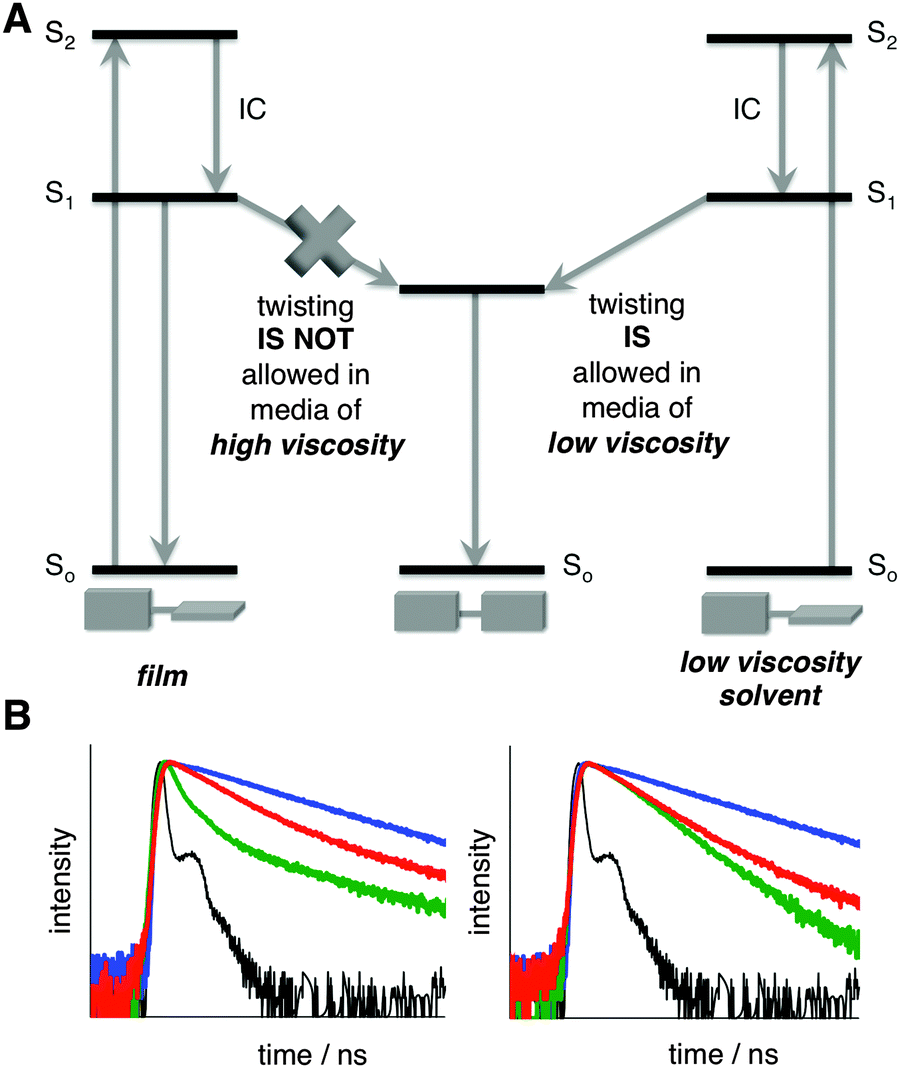 | ||
| Fig. 2 (A) Energy diagram depicting PD excitation, emission, and conformational change in the excited state in media of various viscosities. (B) Normalized time-resolved fluorescent decay traces of PD in EtOH (green), glycerol (red), PVA (blue), and reference (black); λex = 475 nm, λobs = 720 nm (left), λobs = 780 nm (right). See Fig. S3 and Table S1 (ESI†) for additional details. | ||
However, if PD were to be locked in a solid-state matrix (e.g., a polymer), the interconversion between planar and twisted conformations would not be possible. On the other hand, reorganization of the matrix due to external stimuli might have an effect on the conformational bias of PD. Accordingly, we set out to evaluate whether the conformation of PD could be controlled upon subjecting it to mechanical force, i.e., stretching of a polymer film (Fig. S1 and S2, ESI†), such as polyvinyl alcohol (PVA) film, which presents a viable, readily available, and widely used matrix to fix the orientation of various species, including small organic molecules.12
In order to experimentally prove that the ratio between the planar and twisted conformations could be unambiguously established using emission spectra in the solid state, we measured the lifetime of PD in PVA film (where internal rotation is not possible) and compared it to ethanol and glycerol, solvents of low and high viscosity, respectively (Fig. 2B and Fig. S3, Table S1, ESI†). The presence of a negative component in ethanol (Fig. 2B and Table S1, ESI†) demonstrated that the planar conformation received additional “pumping” upon excitation of the twisted conformer,13 thus proving that the rotation in the excited state from the twisted to planar conformation took place. Notably, in glycerol and PVA (Fig. 2B), where the rotation in the excited state is restricted, no negative component was observed.
Next, the steady-state emission of PD was evaluated in the non-stretched (i.e., isotropic) and stretched films (Fig. S2, ESI†). Based on the emission spectra, in the non-stretched film, PD exhibited predominantly the twisted conformation, as judged by the dominant emission peak at ca. 710 nm (Fig. 3 and Fig. S3, ESI†). This observation was not surprising because PD was shown to adopt the twisted conformation in highly viscous media.9b,10b Importantly, progressive stretching of the PVA film (from 5 to 20 mm; Fig. 3A, B and Fig. S5, ESI†) at 70 °C‡ resulted in a significant shift from the twisted to planar conformation, as judged by the increase of the 780/710 nm ratio (Fig. 3). Specifically, the amount of the planar conformation increased from below ca. 40% to 80% (Fig. 3B).
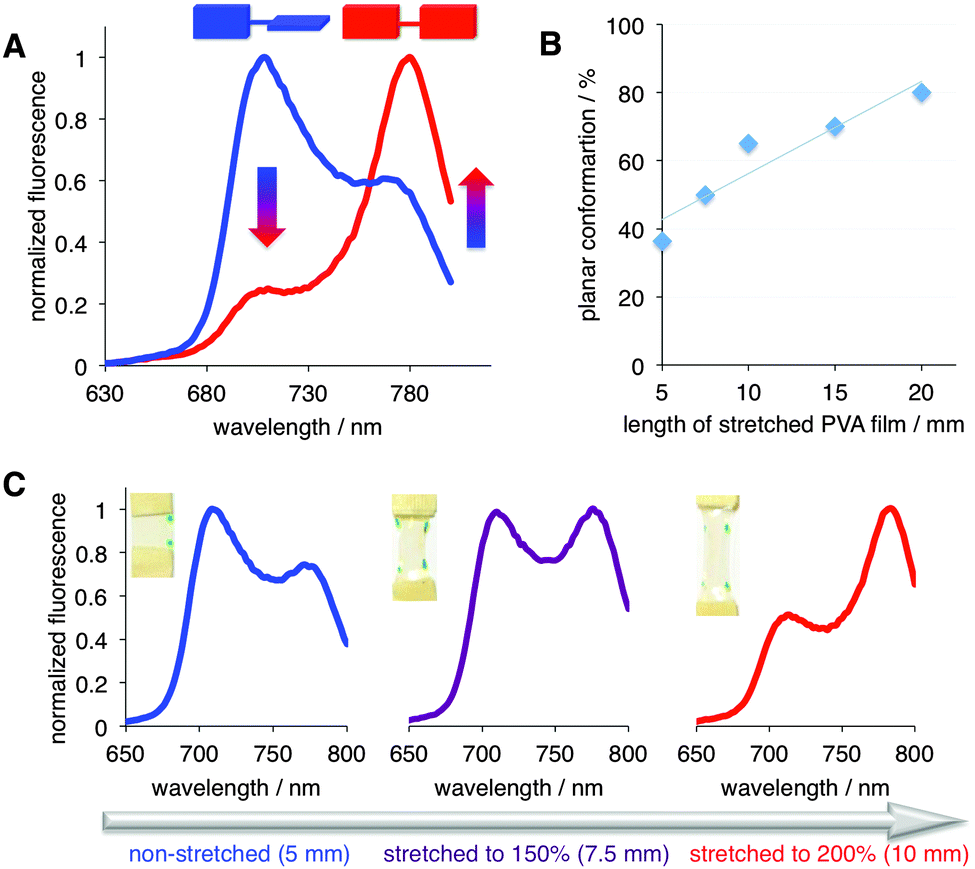 | ||
| Fig. 3 (A) Emission spectra of PD in non-stretched (blue, 5 mm), and stretched to 20 mm, i.e., 400% (red) PVA film. λex = 475 nm (see Fig. S4–S9 (ESI†) for additional details). (B) % of planar conformation of PD as a function of stretching; 5 mm corresponds to the non-stretched film. Line is presented to guide the eye. (C) Stretching-induced conformational transition from twisted (λmaxem ∼ 710 nm) to planar (λmaxem ∼ 780 nm) PD; λex = 475 nm. The dots on the PVA films indicate the extent of stretching (see text and ESI† for additional details). | ||
We also investigated the effect of temperature on the conformation of PD in the non-stretched PVA film (Fig. S6, ESI†). Notably, no significant conformational changes were observed after the isotropic PD/PVA film was heated to 80 °C, as evidenced by the emissions spectra, which indicated that stretching played a crucial part in the conformational change.
The fluorescence spectra were measured under various polarization settings (i.e., both horizontal and vertical polarization planes) in order to rule out any orientation effects induced by stretching of the film, which could have led to a preferential excitation of one conformation by rotating the polarizers (these were placed between the sample and the excitation light as well as between the sample and detector).12a,14 Similar trends were noted under all polarization settings and under a range of excitation wavelengths, i.e., 405, 430, 475, and 510 nm (Fig. S6–S9, ESI†). Importantly, the increase in the planar conformation came at the expense of the twisted conformation regardless of excitation wavelength, i.e., at 405, 430, and 475 nm where the twisted conformation of PD could be excited (Fig. S10, ESI†).
In addition, we examined the possibility that the conformational switch of PD could be due to a change in the degree of crystallinity of the PVA film, since several studies indicated that heating and stretching of PVA films might enhance the crystallinity of the film,15 and it is plausible that the crystalline matrix provides an environment that favors the planar conformation of PD. Fourier-transformed infrared (FTIR) spectroscopy is one of the most convenient spectroscopic tools to estimate the crystallinity of PVA films.16 Analysis of the FTIR spectra of all stretched and non-stretched films revealed that the crystallinity change upon stretching (both under heating and at room-temperature) was less than 5% (Fig. S11, ESI†). Considering that substantial changes in the fluorescence spectra were observed (Fig. 3), it appears that, the morphological changes of the PVA films were not the major contributor to the observed conformational changes of PD. Although the underlying mechanisms for the observed conformational change remain to be clarified, it is evident that mechanothermal stimulation of PD embedded in a polymer matrix could be used to induce and, importantly, preserve a particular conformation of PD. It should be pointed out, that PVA film is not an ideal polymer for reversible stretching; hence the observed results are significant as they strongly indicate that control of the conformation of PD is possible.
Finally, in order to probe whether the conformational change of PD is reversible, we subjected the fully stretched film to heating (Fig. 4).§ As the temperature increased, a shrinkage of the film was noted. Concurrently, an increase in the twisted conformation at the expense of the planar conformation was observed. Notably, when the film heated to 80 °C and was rapidly cooled to 20 °C, the amount of the twisted conformation significantly increased as compared to the initial state (Fig. 4, blue vs. orange traces), while the length of the film decreased.
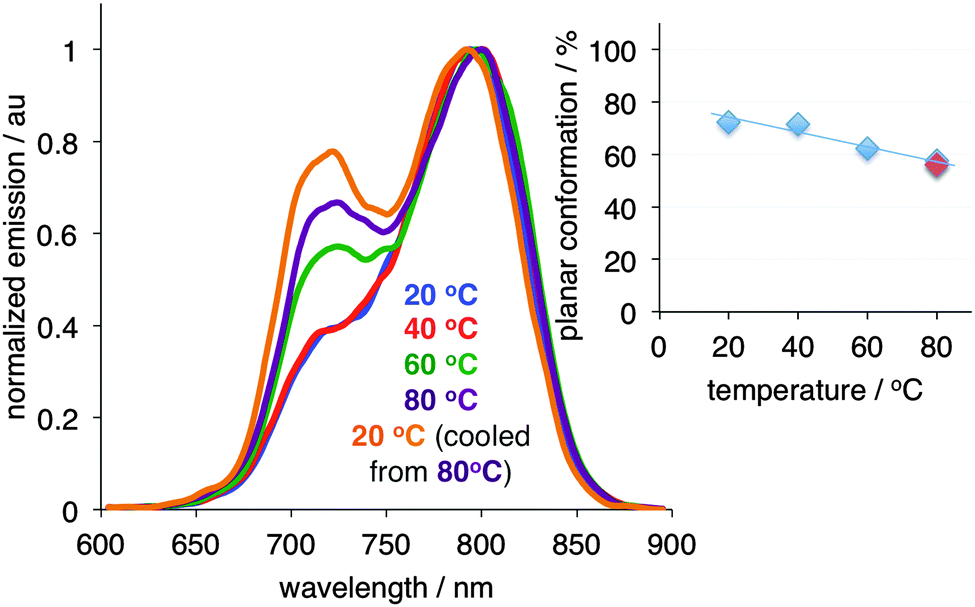 | ||
| Fig. 4 Conformational change of PD upon thermal heating of the stretched (20 mm, i.e., 400% elongation) film in glycerol. Inset: Planar conformation of PD as a function of temperature (light blue symbols correspond to heating of the film from 20 to 40, 60, and 80 °C; light red symbol corresponds to the film being cooled from 80 to 20 °C/orange trace). | ||
Overall, these results revealed that mechanical force could be used to modulate the conformation and optical properties of a porphyrin, which could be a viable strategy for inducing desirable functionalities, with potential applications in materials, sensing, and catalysis. This work was supported in part by NSF (CBET 1403226 to M. B. and S. V. D.) and NIH (R21EB017985 and R01EB12003 to Z. G.). The authors would like to thank Professor Yulia Sevryugina and Mr Arshad Mehmood (TCU-Chemistry & Biochemistry) for access to ATR-FTIR instrument.
Notes and references
- K. C. Kean, S. Akbulatov, Y. Tian, R. A. Widenhoefer, R. Boulatov and S. L. Craig, Angew. Chem., Int. Ed., 2014, 53, 14508–14511 CrossRef PubMed.
- D. Ishikawa, T. Mori, Y. Yonamine, W. Nakanishi, D. L. Cheung, J. P. Hill and K. Ariga, Angew. Chem., Int. Ed., 2015, 54, 8988–8991 CrossRef CAS PubMed.
- S. L. James and T. Friscic, Chem. Soc. Rev., 2013, 42, 7494–7496 RSC ; themed collection: mechanochemistry.
- (a) D. A. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. Van Gogh, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martinez, S. R. White, J. S. Moore and N. R. Sottos, Nature, 2009, 459, 68–72 CrossRef CAS PubMed; (b) G. R. Gossweiler, G. B. Hewage, G. Soriano, Q. Wang, G. W. Welshofer, X. Zhao and S. L. Craig, ACS Macro Lett., 2014, 3, 216–219 CrossRef CAS; (c) X. Fang, H. Zhang, Y. Chen, Y. Lin, Y. Xu and W. Weng, Macromolecules, 2013, 46, 6566–6574 CrossRef CAS; (d) G. Hong, H. Zhang, Y. Lin, Y. Chen, Y. Xu, W. Weng and H. Xia, Macromolecules, 2013, 46, 8649–8656 CrossRef CAS; (e) J. Wang, T. B. Kouznetsova, Z. S. Kean, L. Fan, B. D. Mar, T. J. Martinez and S. L. Craig, J. Am. Chem. Soc., 2014, 136, 15162–15165 CrossRef CAS PubMed; (f) H. Zhang, F. Gao, Z. Cao, Y. Li, Y. Xu, W. Weng and R. Boulatov, Angew. Chem., Int. Ed., 2016, 55, 3040–3044 CrossRef CAS PubMed.
- (a) S. Hou and P. X. Ma, Chem. Mater., 2015, 27, 7627–7635 CrossRef CAS PubMed; (b) N. Hosono, A. M. Kushner, J. Chung, A. R. A. Palmans, Z. Guan and E. W. Meijer, J. Am. Chem. Soc., 2015, 137, 6880–6888 CrossRef CAS PubMed; (c) H.-J. Kim, D. R. Whang, J. Gierschner, C. H. Lee and S. Y. Park, Angew. Chem., Int. Ed., 2015, 54, 4330–4333 CrossRef CAS PubMed; (d) C.-P. Li, J. Chen, C.-S. Liu and M. Du, Chem. Commun., 2015, 51, 2768–2781 RSC; (e) X. Yu, L. Chen, M. Zhang and T. Yi, Chem. Soc. Rev., 2014, 43, 5346–5371 RSC.
- (a) Z. Ma, Z. Wang, M. Teng, Z. Xu and Z. Jia, ChemPhysChem, 2015, 16, 1811–1828 CrossRef CAS PubMed; (b) Y. Sagara, S. Yamne, M. Mitani, C. Weder and T. Kato, Adv. Mater., 2016, 28, 1073–1095 CrossRef CAS PubMed.
- Y. Zhang, K. Wang, G. Zhuang, Z. Xie, C. Zhang, F. Cao, G. Pan, H. Chen, B. Zou and Y. Ma, Chem. – Eur. J., 2015, 21, 2474–2479 CrossRef CAS PubMed.
- (a) M. Levitus, K. Schmeider, H. Ricks, K. D. Shimizu, U. H. F. Bunz and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2001, 123, 4259–4265 CrossRef CAS PubMed; (b) M. Levitus and M. A. Garcia-Garibay, J. Phys. Chem. A, 2000, 104, 8632–8637 CrossRef CAS.
- (a) M. Balaz, H. A. Collins, E. Dahlstedt and H. L. Anderson, Org. Biomol. Chem., 2009, 7, 874–888 RSC; (b) M. K. Kuimova, S. W. Bothchway, A. W. Parker, M. Balaz, H. A. Collins, H. L. Anderson, K. Suhling and P. R. Ogilby, Nat. Chem., 2009, 1, 69–73 CrossRef CAS PubMed; (c) M. K. Kuimova, H. A. Collins, M. Balaz, E. Dahlstedt, J. A. Levitt, N. Segent, K. Suhling, M. Drobizhev, N. S. Makarov, A. Rebane, H. L. Anderson and D. Philips, Org. Biomol. Chem., 2009, 7, 889–896 RSC; (d) H. A. Collins, M. Khurana, E. H. Moriyama, A. Mariampilla, E. Dahlstedt, M. Balaz, M. K. Kuimova, M. Drobizhev, V. X. D. Yang, D. Phillips, A. Rebane, B. C. Wilson and H. L. Anderson, Nat. Photonics, 2008, 2, 420–424 CrossRef CAS.
- (a) L. P. Jameson, M. Balaz, S. V. Dzyuba and N. Kamiya, RSC Adv., 2014, 4, 705–708 RSC; (b) L. P. Jameson, J. D. Kimball, Z. Gryczynski, M. Balaz and S. V. Dzyuba, RSC Adv., 2013, 3, 18300–18304 RSC.
- A. Vyšniauskas, M. Balaz, H. L. Anderson and M. K. Kuimova, Phys. Chem. Chem. Phys., 2015, 17, 7548–7554 RSC.
- (a) A. Mukerjee, T. J. Sorensen, A. P. Ranjan, S. Raut, I. Gryczynski, J. K. Vishwanatha and Z. Gryczynski, J. Phys. Chem. B, 2010, 114, 12679–12684 CrossRef CAS PubMed; (b) Z. Gryczynski, R. Paolesse, K. Smith and E. Bucci, J. Phys. Chem., 1994, 98, 8813–8816 CrossRef CAS; (c) C. Zamora-Ledezma, C. Blanc and E. Anglaret, J. Phys. Chem. C, 2012, 116, 13760–13766 CrossRef CAS; (d) P. Hanczyc, B. Aakerman and B. Norden, Langmuir, 2012, 28, 6662–6669 CrossRef CAS PubMed; (e) F. Di Stasio, P. Kormiychuk, S. Brovelli, P. Usnanski, S. O. McDonnell, G. Winroth, H. L. Anderson, A. Tracz and F. Cacialli, Adv. Mater., 2011, 23, 1855–1859 CrossRef CAS PubMed.
- (a) P. Sarkar, S. V. Koushik, S. S. Vogel, I. Gryczynski and Z. Gryczynski, J. Biomed. Opt., 2009, 14, 034047, DOI:10.1117/1.3156842; (b) R. Ramadass and J. Bereiter-Hahn, J. Phys. Chem. B, 2007, 111, 7681–7690 CrossRef CAS PubMed; (c) B. Zelent, J. M. Vanderkooi, R. Coleman, I. Gryczynski and Z. Gryczynski, Biophys. J., 2006, 91, 3864–3871 CrossRef CAS PubMed.
- (a) J. Goc, A. Dudkowiak, Z. Gryczynski, I. Gryczynski, B. Zelent and F. Frackwiak, J. Fluoresc., 2001, 11, 53–63 CrossRef CAS; (b) A. Dubkowiak, D. Frankowiak, E. Teslak, Z. Gryczynski and I. Gryczynski, Acta Biochim. Pol., 2007, 54, 647–656 Search PubMed.
- (a) K. Kumeta, I. Nagashima, S. Matsui and K. Mizoguchi, Polym. J., 2004, 36, 472–477 CrossRef CAS; (b) K. E. Prasad, B. Das, U. Maitra, U. Ramamurty and C. N. R. Rao, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 13186–13189 CrossRef CAS PubMed; (c) S. K. Sharma, J. Prakash, K. Sudarshah, D. Sen, S. Mazumber and P. K. Pujari, Macromolecules, 2015, 48, 5706–5713 CrossRef CAS.
- (a) N. A. Peppas, Makromol. Chem., 1977, 178, 595–601 CrossRef CAS; (b) O. N. Tretinnikov and S. A. Zagorskaya, J. Appl. Spectrosc., 2012, 70, 521–526 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional spectroscopic characterization of PD as well as the FTIR spectra of PVA films. See DOI: 10.1039/c6cc04306b |
| ‡ 70 °C was chosen to allow for maximum, non-destructive stretching of the PVA film without breakage, while retaining a similar transparency as the non-stretched film as well as preventing subsequent deformation of the film. Stretching the PVA film at room temperature proved to be inefficient: (i) stretching to 10 mm (i.e., 200% elongation) produced a film that underwent shrinkage and deformation; (ii) stretching to 15 mm (i.e., 300% elongation) produced cloudy patches. In both instances, the PVA films were not suitable for spectroscopic measurements. |
| § Heating of the PD-containing PVA film was done in glycerol (solvent in which PVA films are only sparingly soluble) to assure homogeneous heating of the film. |
| This journal is © The Royal Society of Chemistry 2016 |