
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA polarized liquid–liquid interface meets visible light-driven catalytic water oxidation†
Shokoufeh
Rastgar
ab,
Martin
Pilarski
a and
Gunther
Wittstock
 *a
*a
aInstitute of Chemistry, Carl von Ossietzky University of Oldenburg, D-26111 Oldenburg, Germany. E-mail: gunther.wittstock@uni-oldenburg.de; Fax: +49 441 798 3979; Tel: +49 441 798 3971
bHanse-Wissenschaftskolleg, Lehmkuhlenbusch 4, D-27753 Delmenhorst, Germany. Fax: +49 4221 9160 199; Tel: +49 4221 9160 100
First published on 19th August 2016
Abstract
Hyperbranched nanostructured bismuth vanadate at a chemically polarized water/organic interface is applied for efficient visible light-driven catalytic oxidation of water in the presence of [Co(bpy)3](PF6)3 as an organic soluble electron acceptor. The photocurrent response originating from the transfer of photo-excited electrons in BiVO4 to [Co(bpy)3]3+ is measured by scanning electrochemical microscopy.
Inspired by biological photosynthesis, the current research aims at converting sunlight to chemical fuels by driving energetically uphill chemical reactions yielding energy-rich compounds. Conceptually this represents an approach for solving the energy challenge with minimal environmental impact.1–4 Liquid/liquid (L/L) interfaces, or, more precisely, interfaces between two immiscible electrolyte solutions (ITIES), have been proposed as simple models mimicking important functions of biomembranes at which the process of photosynthesis proceeds in green plants. The analogy extends to charge carrier separation by two interacting photocenters (equivalent to a z-scheme) and catalyst regeneration at interfaces.5 ITIES can be polarized either externally using a potentiostat or chemically by changing the composition or concentration of supporting electrolytes in the organic or aqueous phase. The applied galvanic potential difference across the interface could control the efficiency and pathway of interfacial processes involving charged reactants.6–8 Hence, the design of efficient artificial photosynthesis systems based on charge transfer at ITIES has recently received significant attention.9–11 Such systems utilize sunlight to split water into molecular oxygen O2 and hydrogen H2 (as renewable solar fuels).11 The water oxidation reaction (WOR), a multi-electron multi-proton transfer, is energy demanding and considered as the most challenging step in water splitting. It requires suitable catalytic conditions for reasonable rates and/or overpotentials.12
Bismuth vanadate (BiVO4) is one of the most interesting visible-light-driven photocatalysts for water oxidation, because it has a suitable position of the valence band (VB) edge at ca. 2.4 eV vs. RHE, a sufficiently narrow bandgap for visible light absorption, is stable, abundant and of low cost. However, it suffers from poor charge transport properties causing excessive electron–hole recombination.13–17 Furthermore, the hole transfer kinetics for WOR is sluggish.14 One direction for further improvement is size and shape control in order to facilitate the collection and separation of electron–hole pairs at the semiconductor/solution interfaces.14
Our approach herein is the use of hyperbranched BiVO4 at a chemically polarized ITIES18 for enhancing the rate of photocatalytic WOR (Fig. 1). The nanoscale branches minimize the charge carrier diffusion length to the interface where they are converted before recombining. This is further supported by [Co(bpy)3](PF6)3 as an electron-acceptor in the organic phase. By efficient relaying of the photo-excited electrons from BiVO4 to the electron acceptor the electron/hole pair separation is facilitated after photoexcitation in BiVO4. This function is analogous to that of the electron transport chain between photosystems I and II.1 Interestingly, the driving force for electron transfer to [Co(bpy)3]3+ can be controlled easily by changing the polarization of the ITIES, which then clearly influences the efficiency of electron transfer from water to photogenerated holes (yielding O2).
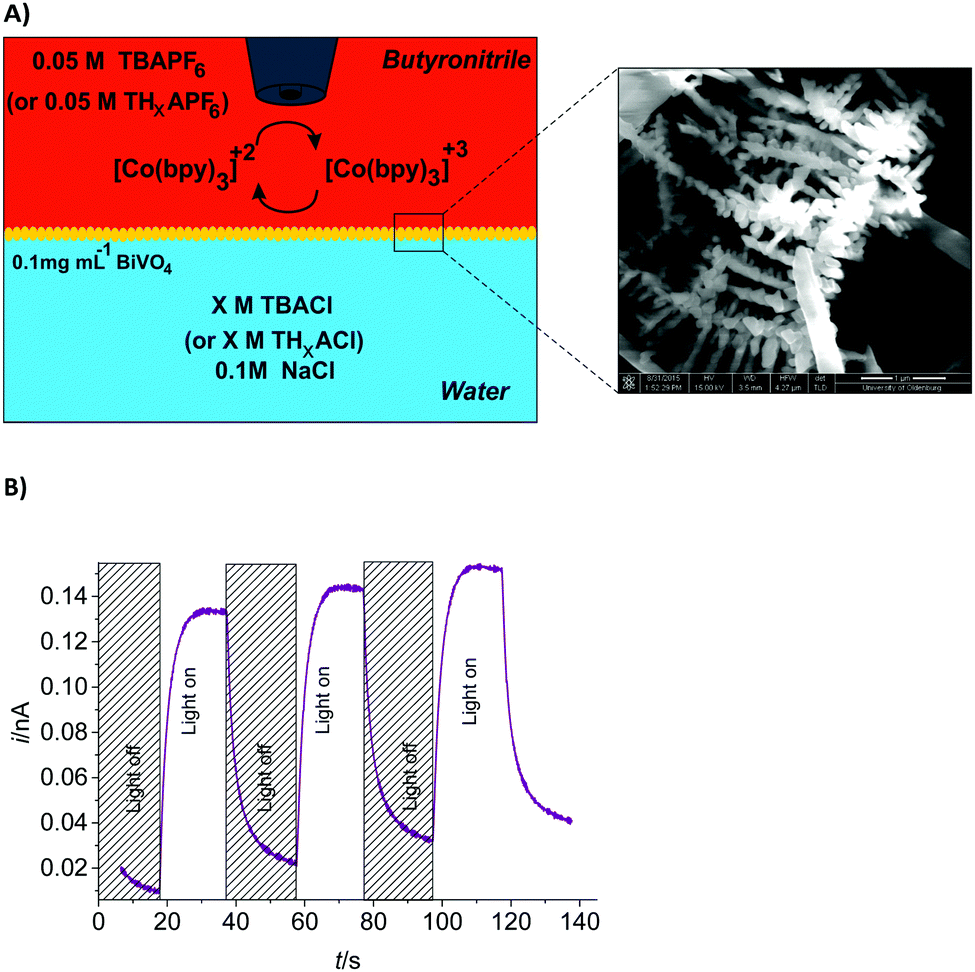 | ||
| Fig. 1 (A) Schematic representation of the initial composition of the aqueous and organic phases for chemical polarization of the L/L interface used for the study of BiVO4 photoactivity. Magnified SEM image of the hyperbranched structure of BiVO4; (B) typical photocurrent transient for the detection of [Co(bpy)3]2+ recorded at a Au ME (rT = 12.5 μm, d = 20 μm) with an applied potential of ET = 0.5 V (vs. Ag quasi-reference electrode (AgQRE)). The butyronitrile phase contained 0.5 mM [Co(bpy)3](PF6)3 and 0.05 M TBAPF6, and the aqueous phase contained 0.1 M NaCl and 0.01 M TBACl. | ||
In the present work, the interfacial photo-induced electron transfer (ET) reaction is studied by means of scanning electrochemical microscopy (SECM), which has quickly become an important tool for probing rapid processes at ITIES, including ET,19–22 ion transfer,23–26 and molecular transfer27 with high sensitivity. SECM is used here for recording the scavenging of photoelectrons by reduction of [Co(bpy)3]3+ at the BiVO4/butyronitrile interface. The SECM microelectrode (ME) oxidizes the resulting [Co(bpy)3]2+. Finally, WOR by photo-generated holes of BiVO4 at the chemically polarized ITIES is followed by online detection of O2 while the cell is irradiated using visible light (λ > 420 nm).
The composition of the cell with the ITIES is schematically shown in Fig. 1A. Tetrabutylammonium chloride (TBACl) and tetrabutylammonium hexafluorophosphate (TBAPF6) are the supporting electrolytes in water and butyronitrile phases, respectively. Tetrabutylammonium (TBA+) is highly hydrophobic and acts as a common ion which induces the specific potential difference across the interface according to the Nernst–Donnan equation.22,28
The BiVO4 nanocrystals are initially well-dispersed in the aqueous phase. The ITIES polarized immediately by ion-transfer and then BiVO4 nanocrystals are driven electrostatically to the interface. The synthesized BiVO4 nanocrystals resemble the shape of trunks with nano-scaled branches (so-called hyper-branched structure). Detailed data on the synthesis and characterization of BiVO4 and [Co(bpy)3]2+/3+ are provided in the ESI.†
Photoinduced ET from nanocrystalline BiVO4 to [Co(bpy)3]3+ is investigated using SCEM. A Au ME with radius rT = 12.5 μm is placed in the upper, organic phase at a distance d = 20 μm to the ITIES at a potential ET of +0.5 V (vs. Ag quasi-reference electrode (AgQRE)) to detect sensitively [Co(bpy)3]2+ formed by the photoinduced ET from BiVO4 to [Co(bpy)3]3+ before it is diluted in the bulk of the organic phase. The Au ME is approached by monitoring the hindered diffusion of [Co(bpy)3]3+ in the organic phase. The transient [Co(bpy)3]2+ oxidation current at the Au ME is shown in Fig. 1B. No photoresponses are detected if either BiVO4, [Co(bpy)3]3+ or illumination is absent (data not shown).
Clearly, band-gap illumination of the interface leads to interfacial ET to [Co(bpy)3]3+ in the organic phase. The generated photo-induced electron–hole pairs with quasi-Fermi levels approach the energies of the conduction band (CB) and valence band (VB) of BiVO4. The CB energy of BiVO4 is +0.02 V29 and the redox potential of [Co(bpy)3]3+ in butyronitrile is +0.34 V vs. RHE. Therefore, heterogeneous ET of CB electrons from BiVO4 to [Co(bpy)3]3+ appears thermodynamically feasible.
The driving force of the interfacial photo-induced ET reaction is determined by this thermodynamic energy difference and the galvanic potential difference (Δorgaoϕ) across the ITIES.30,31 The influence of Δorgaoϕ is investigated by adjusting it by means of the concentration ratio of TBA+ between the aqueous and organic phases. Cyclic voltammograms (CVs) of [Co(bpy)3]3+ reduction were recorded in the butyronitrile solution as part of a biphasic system containing 0.01–0.1 M TBA+ in the aqueous phase and a fixed concentration of 0.05 M TBA+ in the organic phase. As shown in the inset of Fig. 2, the reduction potential of [Co(bpy)3]3+ shifts positively by nearly 60 mV per concentration decade of TBA+, in good agreement with the Nernst–Donnan equation. Please note that the electroneutrality of the two phases is maintained by insertion of the hydrophobic, potential-determining ion TBA+ into the organic phase. In addition, the influence of Δorgaoϕ is observed clearly by photocurrent responses at the Au ME which grows with the TBA+ concentration in the aqueous phase (Fig. 2). This causes a more positive potential across the interface contributing to a higher driving force for photo-induced ET.
 | ||
| Fig. 2 Photocurrent transients for the detection of [Co(bpy)3]2+ in butyronitrile at Au ME (rT = 12.5 μm, ET = +0.5 V (vs. AgQRE)) with different concentrations of TBACl in the aqueous phase. The butyronitrile solution contained 0.5 mM [Co(bpy)3](PF6)3 and 0.05 M TBAPF6. From bottom to top, the aqueous solution contained 0.01, 0.025, 0.05, and 0.1 M TBACl. The inset figure is the dependence of the half-wave potential (E1/2) on TBA+ concentration. | ||
Interestingly, changing the aqueous electrolyte from acidic to alkaline media dramatically influences the photocurrent responses. As depicted in Fig. 3, the transient signal increases significantly in neutral (pH 7, 0.1 M NaCl) or alkaline (pH 9, 0.01 mM NaOH) aqueous electrolytes compared to the acidic solution (pH 3, 0.02 M HClO4).
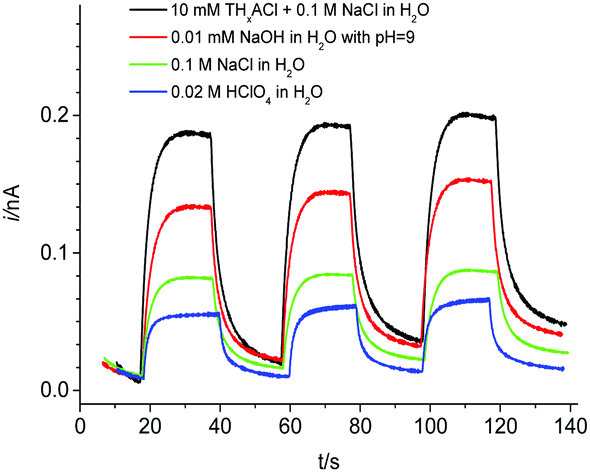 | ||
| Fig. 3 Photocurrent transients for the detection of [Co(bpy)3]2+ in butyronitrile at a Au ME (rT = 12.5 μm, ET = +0.5 V (vs. AgQRE)) with different aqueous supporting electrolytes. The butyronitrile solution contained 0.5 mM [Co(bpy)3](PF6)3 and 0.05 M TBAPF6. The aqueous solution contained 0.1 M NaCl and additionally from bottom to top, 0.02 M HClO4, no addition, 0.01 mM NaOH and 0.01 M THxACl. | ||
It is known that the energy levels of the CB and VB of the BiVO4 semiconductor shift with pH, whereas the redox potential of [Co(bpy)3]3+/2+ is independent of pH.32 A higher pH of the reactant solution yields a negative shift of the band-levels of BiVO4 (on the electrochemical scale). Consequently, the CB electrons in BiVO4 attain more reductive power towards the approximately pH-independent formal potential of [Co(bpy)3]3+ and the photo-induced ET proceeds more easily (Fig. 3). Furthermore, the photocurrent signal is enhanced when using tetrahexylammonium (THxA+) instead of TBA+ as the common ion because the more hydrophobic THxA+ has a higher tendency to transfer to the organic phase and, therefore, contributes to a higher driving force for the photo-induced ET reaction from BiVO4 to [Co(bpy)3]3+.18
Parallel to the ET reaction between photo-excited electrons of BiVO4 and [Co(bpy)3]3+, photo-generated holes in the VB of BiVO4 cause the WOR which is energetically possible. The reaction product, O2, can be detected by online mass spectrometry (ESI-6, ESI†) as the main product of photooxidation of water. Table 1 summarizes the O2 evolution rates per gram catalyst within the first 10 min of irradiation. The O2 evolution rate is improved drastically under polarization of the ITIES with TBA+ compared to batches without either potential-determining common ions or [Co(bpy)3](PF6)3 as an electron acceptor in the organic phase. By trapping the photo-excited electrons, the Co complex facilitates both charge separation in BiVO4 and efficient O2 production at the BiVO4/water interface. Furthermore, the increased driving force due to the polarization of the ITIES by a common ion increases the O2 evolution rate.
| Batcha | [Salt]aqb | [Co(bpy)33+]orgc | O2 evolution rated/(μmol h−1 g−1) |
|---|---|---|---|
| a Details of the photoreactor and GC detection in ESI-6, ESI. b 30 mg BiVO4 was dispersed in 35 mL water. c 35 mL organic phase contained 0.05 M TBAPF6. d Per gram of catalyst. e Hyperbranched BiVO4. f Powdered catalyst synthesized by the sol–gel method.41,42 | |||
| 1e | None | 0 | 0 |
| 2e | 0.1 M NaCl + 0.01 TBACl | 0 | 5 |
| 3e | None | 0.5 mM | 218 |
| 4e | 0.01 M NaCl + 0.01 M TBACl | 0.5 mM | 336 |
| 5f (powder) | 0.1 M NaCl + 0.01 TBACl | 0.5 mM | 0 |
The removal of the electrons from the BiVO4 nanostructures inhibits the otherwise fast recombination of electron–hole pairs. This requires nanostructured BiVO4 for efficient transport of electrons to the solid–liquid interface. When using the same mass of powdered BiVO4 without hyperbranched structures, the O2 evolution rate is below the detection limit. So far, many strategies have been developed to increase the efficiency of visible light-driven oxygen evolution at BiVO4 semiconductor materials applied as photocatalyst powders32–39 or modified solid electrodes16,17 in aqueous solutions with electron acceptors such as Co32 or Fe34,40 complexes, morphology control33 and facet engineering35 of BiVO4-based materials. Here, the extremely high rate of visible light-driven O2 evolution allows detection within only 10 min of illumination of the hyperbranched photocatalyst powder by separation of photo-induced charge carriers at a molecular interface. A similar value of 238 μmol h−1 g−1 is also obtained with Ag+ as a scavenger in the first 15 min of operation and with a larger optical band (λ > 320 nm).
The very high initial activity decreased within 20–30 min to levels below the detection limit of our system (ca. 1 μmol h−1). The data are shown in Fig. 4. The accumulated amounts of O2 generated during the reaction time are plotted in Fig. 4. They compare favourably to amounts of O2 detected within 1–5 hours for BiVO4 systems as reported recently.32–39
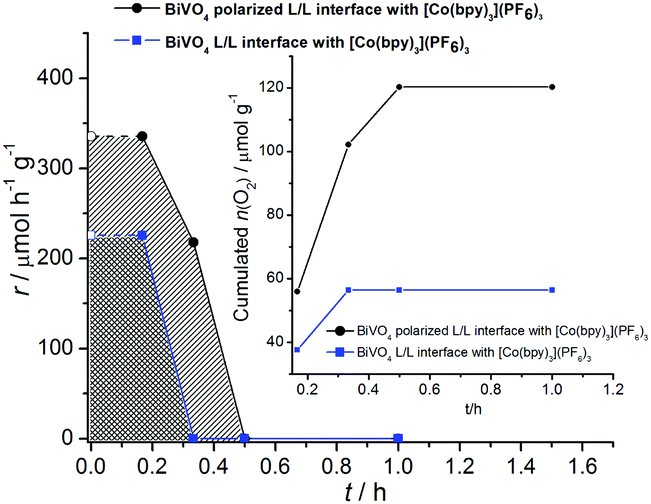 | ||
| Fig. 4 Oxygen evolution rate over the reaction time. The inset figure is the cumulated amount of generated O2 assuming a linear change of O2 concentration in the exhaust gas stream between the sampling. The initial oxygen production rate at t = 0 is assumed to be the same as that at the first sampling after 10 min; (●) hyperbranched BiVO4, 0.01 M TBACl aqueous phase, 0.5 mM [Co(bpy)3](PF6)3 + 0.05 M TBAPF6, (■) hyperbranched BiVO4, aqueous phase without TBACl, 0.5 mM [Co(bpy)3](PF6)3 + 0.05 M TBAPF6. | ||
In conclusion, a completely new approach has been proposed by which the photochemical behavior of BiVO4 semiconductor nanostructures can be improved for visible light-driven oxidation of water. The nanoparticulate BiVO4 semiconductor is assembled at the water/butyronitrile interface polarized by the common, highly hydrophobic tetraalkylammonium cation partitioning between the two liquid phases. A CoIII complex acts as an electron acceptor in the organic phase. The high surface area of nanosized BiVO4 crystals with a specific hyperbranched structure along with the defect free ITIES minimizes the interfacial recombination of photo-excited electron–hole pairs by rapidly transferring electrons to reactants in the adjacent liquid phases. The ET reaction can be characterized sensitively by oxidation of [Co(bpy)3]2+ originating from the photo-driven reaction at the ITIES using a SECM configuration in the organic phase.
Moreover, the driving force for the photo-induced ET reaction could be readily maximized by chemically controlling the polarization of the interface. This improves the WOR compared to recent reports on WOR using powdered BiVO4 photocatalysts. It is expected that the new strategy developed in this work will promote efficient water splitting with powdered BiVO4 photocatalysts in large-scale solar-to-hydrogen conversion. Further studies will be directed to address details of WOR mechanisms at BiVO4 at ITIES, integrating other approaches such as doping of BiVO4.
We are grateful to Prof. Dr Michael Wark and Sven Warfsmann (Institute of Chemistry, Carl von University of Oldenburg) for help with measuring O2 evolution rates. S. R. thanks the Hanse Institute of Advanced Studies Delmenhorst and the Alexander von Humboldt Foundation for research fellowships.
Notes and references
- M. K. Dymond, C. V. Hague, A. D. Postle and G. S. Attard, J. R. Soc., Interface, 2013, 10, 1 Search PubMed.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446 CrossRef CAS PubMed.
- M. D. Kärkäs, O. Verho, E. V. Johnston and B. r. Åkermark, Chem. Rev., 2014, 114, 11863 CrossRef PubMed.
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Chem. Rev., 2012, 112, 4016 CrossRef CAS PubMed.
- R. Lahtinen, D. J. Fermın, K. Kontturi and H. H. Girault, J. Electroanal. Chem., 2000, 483, 81 CrossRef CAS.
- N. Eugster, D. J. Fermín and H. H. Girault, J. Phys. Chem. B, 2002, 106, 3428 CrossRef CAS.
- N. Eugster, D. J. Fermín and H. H. Girault, J. Am. Chem. Soc., 2003, 125, 4862 CrossRef CAS PubMed.
- H. Nagatani, S. Dejima, H. Hotta, T. Ozeki and T. Osakai, Anal. Sci., 2004, 20, 1575 CrossRef CAS PubMed.
- J. K. Cooper and I. Benjamin, J. Phys. Chem. B, 2014, 118, 7703 CrossRef CAS PubMed.
- D. Schaming, I. Hatay, F. Cortez, A. Olaya, M. A. Méndez, P. Y. Ge, H. Deng, P. Voyame, Z. Nazemi and H. Girault, Chimia, 2011, 65, 356 CrossRef CAS PubMed.
- H. Inoue, T. Shimada, Y. Kou, Y. Nabetani, D. Masui, S. Takagi and H. Tachibana, ChemSusChem, 2011, 4, 173 CAS.
- A. Kudo, K. Omori and H. Kato, J. Am. Chem. Soc., 1999, 121, 11459 CrossRef CAS.
- J. Gan, X. Lu and Y. Tong, Nanoscale, 2014, 6, 7142 RSC.
- Z.-F. Huang, L. Pan, J.-J. Zou, X. Zhang and L. Wang, Nanoscale, 2014, 6, 14044 RSC.
- Y. Park, K. J. McDonald and K.-S. Choi, Chem. Soc. Rev., 2013, 42, 2321 RSC.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520 RSC.
- A. G. Volkov, D. W. Deamer, D. L. Tanelian and V. S. Markin, Liquid Interfaces in Chemistry and Biology, J Wiley, New York, 1998 Search PubMed.
- F. Li, B. Su, F. C. Salazar, R. P. Nia and H. H. Girault, Electrochem. Commun., 2009, 11, 473 CrossRef CAS.
- P. Sun, F. Li, Y. Chen, M. Zhang, Z. Zhang, Z. Gao and Y. Shao, J. Am. Chem. Soc., 2003, 125, 9600 CrossRef CAS PubMed.
- Z. Ding, B. M. Quinn and A. J. Bard, J. Phys. Chem. B, 2001, 105, 6367 CrossRef CAS.
- F. Li and P. R. Unwin, J. Phys. Chem. C, 2015, 119, 4031 CAS.
- R. Ishimatsu, J. Kim, P. Jing, C. C. Striemer, D. Z. Fang, P. M. Fauchet, J. L. McGrath and S. Amemiya, Anal. Chem., 2010, 82, 7127 CrossRef CAS PubMed.
- X. Lu, T. Wang, X. Zhou, Y. Li, B. Wu and X. Liu, J. Phys. Chem. C, 2011, 115, 4800 CAS.
- Y. Wang, K. Kececi, J. Velmurugan and M. V. Mirkin, J. Chem. Sci., 2013, 4, 3606 CAS.
- S. Amemiya and A. J. Bard, Anal. Chem., 2000, 72, 4940 CrossRef CAS PubMed.
- J. Strutwolf, J. Zhang, A. L. Barker and P. R. Unwin, Phys. Chem. Chem. Phys., 2001, 3, 5553 RSC.
- W. Adamiak, J. Jedraszko, O. Krysiak, W. Nogala, J. C. Hidalgo-Acosta, H. H. Girault and M. Opallo, J. Phys. Chem. C, 2014, 118, 23154 CAS.
- S. J. Hong, S. Lee, J. S. Jang and J. S. Lee, Energy Environ. Sci., 2011, 4, 1781 CAS.
- M. Tsionsky, A. J. Bard and M. V. Mirkin, J. Phys. Chem. C, 1996, 100, 17881 CrossRef CAS.
- H. Jensen, D. J. Fermín, J. E. Moser and H. H. Girault, J. Phys. Chem. B, 2002, 106, 10908 CrossRef CAS.
- Y. Sasaki, H. Kato and A. Kudo, J. Am. Chem. Soc., 2013, 135, 5441 CrossRef CAS PubMed.
- Y. Zhao, Y. Xie, X. Zhu, S. Yan and S. Wang, Chem. – Eur. J., 2008, 14, 1601 CrossRef CAS PubMed.
- D. J. Martin, P. J. T. Reardon, S. J. A. Moniz and J. Tang, J. Am. Chem. Soc., 2014, 136, 12568 CrossRef CAS PubMed.
- D. Wang, H. Jiang, X. Zong, Q. Xu, Y. Ma, G. Li and C. Li, Chem. – Eur. J., 2011, 17, 1275 CrossRef CAS PubMed.
- P. Zhou, J. Yu and M. Jaroniec, Adv. Mater., 2014, 26, 4920 CrossRef CAS PubMed.
- A. Iwase, Y. H. Ng, Y. Ishiguro, A. Kudo and R. Amal, J. Am. Chem. Soc., 2011, 133, 11054 CrossRef CAS PubMed.
- Y. Sasaki, H. Nemoto, K. Saito and A. Kudo, J. Phys. Chem. C, 2009, 113, 17536 CAS.
- Q. Jia, A. Iwase and A. Kudo, J. Chem. Sci., 2014, 5, 1513 CAS.
- M. T. McDowell, M. F. Lichterman, J. M. Spurgeon, S. Hu, I. D. Sharp, B. S. Brunschwig and N. S. Lewis, J. Phys. Chem. C, 2014, 118, 19618 CAS.
- P. Pookmanee, S. Kojinok and S. Phanichphant, J. Met., Mater. Miner., 2012, 22, 49 CAS.
- O. Merka, O. Raisch, F. Steinbach, D. W. Bahnemann and M. Wark, J. Am. Ceram. Soc., 2013, 96, 634 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc04275a |
| This journal is © The Royal Society of Chemistry 2016 |