
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective dual-purpose photocatalysis for simultaneous H2 evolution and mineralization of organic compounds enabled by a Cr2O3 barrier layer coated on Rh/SrTiO3†
Young-Jin
Cho
a,
Gun-hee
Moon
a,
Tomoki
Kanazawa
b,
Kazuhiko
Maeda
b and
Wonyong
Choi
*a
aSchool of Environmental Science and Engineering, Pohang University of Science and Technology (POSTECH), Pohang, 790-784, Republic of Korea. E-mail: wchoi@postech.edu
bDepartment of Chemistry, School of Science, Tokyo Institute of Technology, 2-12-1-NE-2 Ookayama, Meguro-ku, Tokyo 152-8550, Japan
First published on 28th June 2016
Abstract
Dual-functional photocatalysis for H2 evolution with the simultaneous mineralization of 4-chlorophenol was achieved under de-aerated conditions using a Cr2O3/Rh/SrTiO3 photocatalyst which has Rh nanoparticles covered with a thin Cr2O3 barrier layer to selectively control and maximize the dual-functional photocatalytic activity.
Hydrogen is considered as an ideal energy storage medium and a promising energy carrier since it can be obtained from abundant natural resources such as water and biomass instead of fossil fuels.1–4 In particular, photocatalytic water splitting is widely studied as a promising technology to produce hydrogen using solar light.5–7 Another important application of photocatalysis is the degradation of organic compounds for the remediation of polluted water and air.8–12 In photocatalytic H2 production, organic electron donors are commonly used as sacrificial agents to scavenge photogenerated holes.13–17 However, the intentional addition of organic electron donors (e.g., alcohols, organic acids, amines) for H2 production is not practically acceptable since the electron donors themselves are another energy resource (often more expensive than H2). Therefore, a more desirable strategy is to use organic waste and pollutants in water as in situ electron donors. This is the concept of dual-functional photocatalysis which produces H2 along with the simultaneous degradation of organic pollutants.18–20 Recently, Kim et al. demonstrated that the simultaneous H2 production with the anoxic photocatalytic degradation of organic compounds can be achieved by using TiO2 modified by both surface fluorination and Pt deposition (F-TiO2/Pt).19,20 However, the total organic carbon (TOC) removal efficiency was negligible and TOC remained almost unchanged during the photocatalytic degradation of 4-chlorophenol (4-CP) on F-TiO2/Pt, because dioxygen was needed for mineralization. Meeting the optimal condition for dual-purpose photocatalysis is contradictory because the H2 evolution requires anoxic conditions whereas the mineralization of organic compounds needs dioxygen.
In this study, a selective dual-purpose photocatalysis that achieves H2 production and TOC removal simultaneously under anoxic conditions is reported. Instead of using the combination of TiO2 (as a base photocatalyst) and Pt (as a cocatalyst for H2 evolution), SrTiO3 (base photocatalyst) and Rh@Cr2O3 core–shell nanostructure (cocatalyst) were employed in this work. The photocatalytic activities of the composite materials of Cr2O3/Pt/SrTiO3 and other Rh@Cr2O3 (core–shell) loaded metal oxides have been previously demonstrated for the overall water splitting.21,22 In this work, we demonstrate that Cr2O3/Rh/SrTiO3 can be a promising dual-purpose photocatalyst that can produce H2 along with TOC removal (mineralization) of aromatic pollutants in a de-aerated aqueous suspension which is considered an inappropriate condition to achieve the mineralization of organic pollutants.
Cr2O3/Rh/SrTiO3 photocatalyst was prepared by step-wise photo-deposition of Rh nanoparticles on the SrTiO3 surface as a core and then the Cr2O3 nano-shell on the Rh core.22 The loading amount of Rh and Cr2O3 was 0.5 wt% and 0.75 wt%, respectively. Fig. S1 (ESI†) shows the HRTEM images of Cr2O3/Rh/SrTiO3 and Rh/SrTiO3 photocatalysts. Rh nanoparticles of 2–4 nm diameter were observed to be deposited on the surface of both photocatalysts and the Cr2O3 shell was seen around the Rh core of the Cr2O3/Rh/SrTiO3 catalyst. Rh/SrTiO3 and Cr2O3/Rh/SrTiO3 were compared for the photocatalytic degradation of 4-CP under de-aerated conditions as shown in Fig. 1. Cr2O3/Rh/SrTiO3 exhibited a much higher activity than Rh/SrTiO3 in both the removal of 4-CP and the concurrent production of chloride (Fig. 1a). The TOC removal was also highly enhanced with Cr2O3/Rh/SrTiO3 (Fig. 1b). These results clearly indicate that the removal of 4-CP in the suspension of Cr2O3/Rh/SrTiO3 proceeded along with the mineralization (i.e., TOC removal) even under the de-aerated conditions, where the initial dissolved O2 was measured to be less than 0.1 ppm using a dissolved O2 meter. In the absence of O2 that serves as a main electron scavenger, the photocatalytic oxidation and mineralization should be inhibited on a bare semiconductor but the loading of Rh and Cr2O3 changes the photocatalytic reaction mechanism.
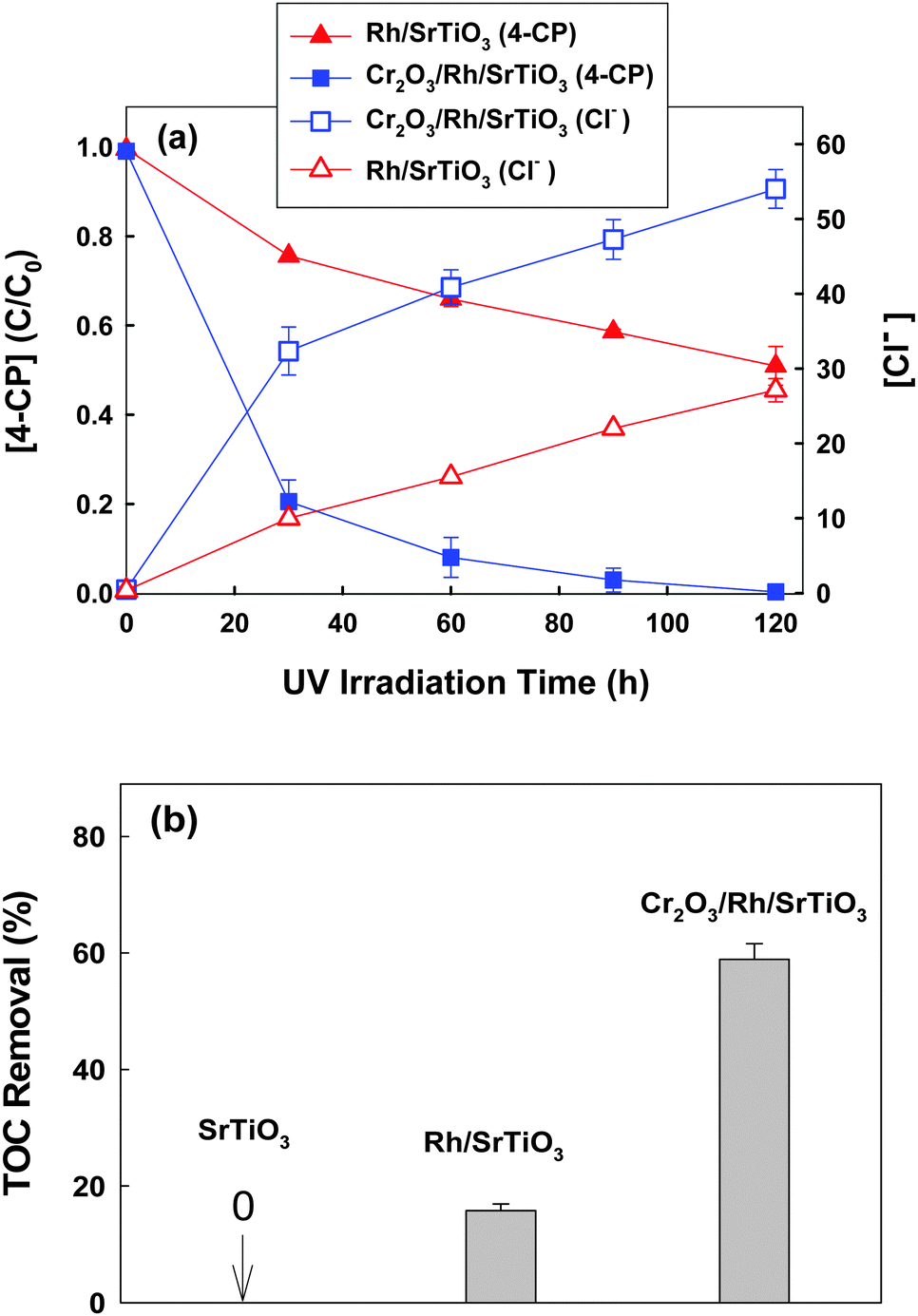 | ||
| Fig. 1 (a) Photocatalytic degradation of 4-CP and the concurrent production of chloride in a de-aerated catalyst suspension. (b) Comparison of TOC removal efficiencies after 2 h photocatalytic reaction. Experimental conditions: [catalyst] = 0.5 g L−1, [4-CP]0 = 100 μM, pH0 = 7, λ > 320 nm, air-tight, and initially Ar-purged for 1 h before UV irradiation. | ||
H2 evolution was also monitored during the anoxic degradation of 4-CP. As seen in Fig. 2a, markedly increased H2 production was observed with the Cr2O3/Rh/SrTiO3 photocatalyst compared with Rh/SrTiO3. The initial rate of H2 production on Cr2O3/Rh/SrTiO3 was around 12 μmol h−1. The apparent photonic efficiency of H2 evolution (with 300 μM 4-CP) was separately measured under the irradiation centered around λ = 330 ± 10 nm and determined to be 0.7%. Concurrent O2 evolution was also observed in the case of Cr2O3/Rh/SrTiO3, whereas no O2 production was observed with Rh/SrTiO3. Such a difference can be ascribed to the fact that the Cr2O3 layer over Rh can suppress the back reaction of H2 with O2 to produce H2O.23 The fact that O2 was evolved along with the degradation of 4-CP on Cr2O3/Rh/SrTiO3 indicates that holes react with both 4-CP and water molecules. The in situ generated O2 should be subsequently consumed during the mineralization of 4-CP,19,24,25 which explains why the degradation of 4-CP was possible under de-aerated conditions. As a result, the in situ O2 evolution was significantly enhanced in the absence of 4-CP (Fig. 2b). The mineralization of 4-CP can be expressed by eqn (1).
| C6H5OCl (6Corg) + 6.5O2 → 6CO2 + 2H2O + HCl | (1) |
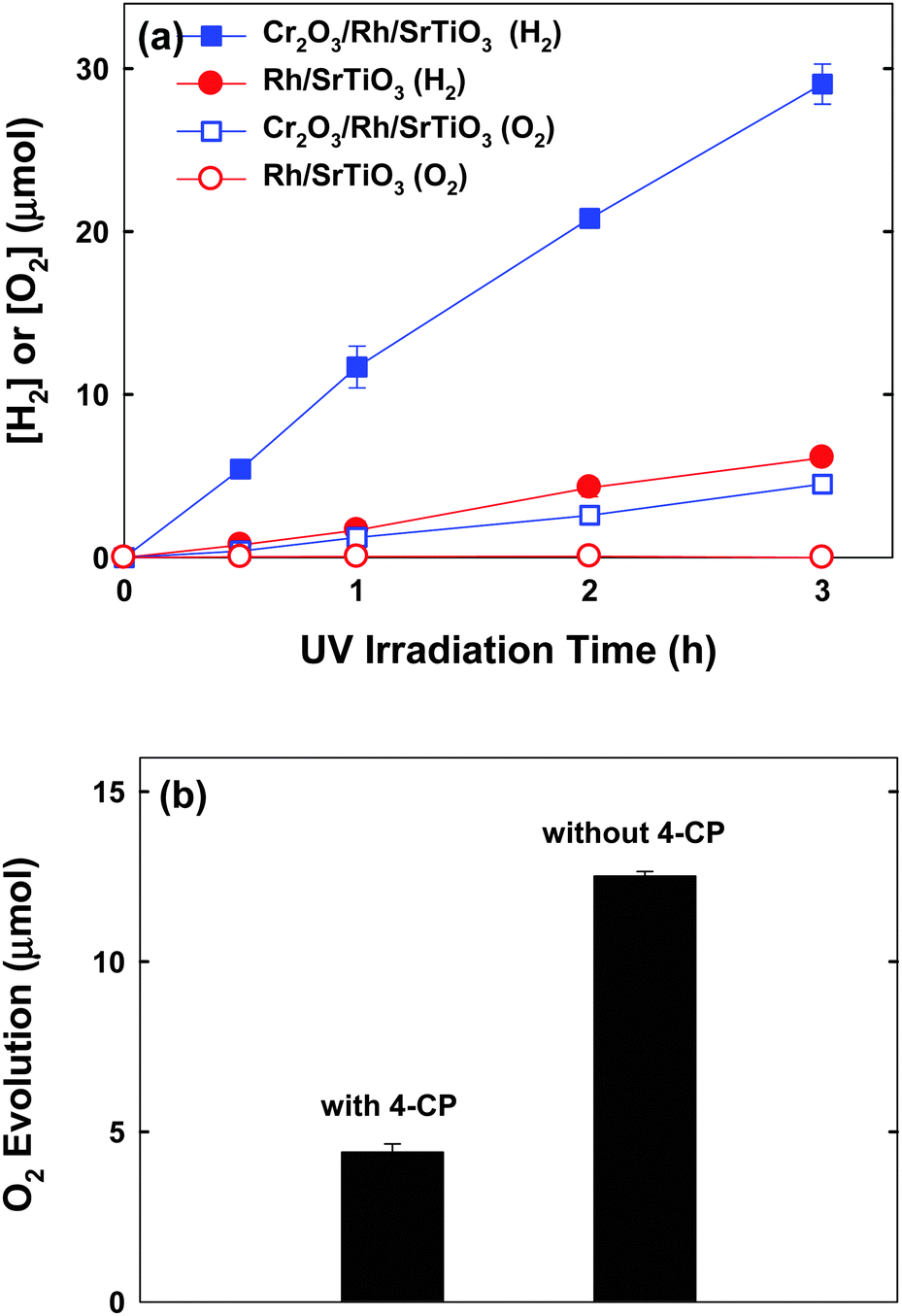 | ||
| Fig. 2 (a) Time-profiled production of H2 and O2 in the suspension of Cr2O3/Rh/SrTiO3 and Rh/SrTiO3 with 4-CP (300 μM) in the initially de-aerated suspension. (b) Comparison of photocatalytic O2 evolution in the suspension of Cr2O3/Rh/SrTiO3 (after 3 h reaction) in the presence and absence of 4-CP. | ||
The effects of dissolved O2 and the probe reagents (i.e., t-butyl alcohol (TBA) and EDTA) were further investigated to understand the photocatalytic mechanisms of Cr2O3/Rh/SrTiO3 and Rh/SrTiO3, as shown in Fig. S3 (ESI†). First, it is noted that the effects of dissolved O2 are drastically different between Cr2O3/Rh/SrTiO3 and Rh/SrTiO3. The presence and absence of O2 did not affect the 4-CP degradation on Cr2O3/Rh/SrTiO3 at all while the degradation of 4-CP on Rh/SrTiO3 was significant only in the presence of O2. This observation is fully consistent with the previous reports that the Cr2O3 shell layer covering the Rh core blocks the contact of O2 with the Rh core, but is still permeable to protons.22,23 As a result, the CB electron transfer to O2 on Cr2O3/Rh/SrTiO3 is insignificant but the CB electron transfer to protons (H+) is allowed with enabling the concurrent hole transfer to 4-CP. Fig. S3 (ESI†) also shows that the photocatalytic degradation of 4-CP remains unchanged in the presence of excessive TBA (as an OH radical scavenger) but is highly retarded in the presence of excessive EDTA (as a hole scavenger).26 This supports that 4-CP degradation proceeds via a direct hole-transfer, and not an OH radical-meditated pathway.
To investigate the role of the Cr2O3 shell on the Rh core and its effects on the interfacial electron transfer on Cr2O3/Rh/SrTiO3 and Rh/SrTiO3, the Fe3+/2+ redox couple-mediated photocurrent was collected (via reactions (2) and (3)) in the UV-irradiated suspension of each catalyst.27
| Fe3+ + ecb− → Fe2+ (on photocatalyst) | (2) |
| Fe2+ → Fe3+ + e− (on Pt electrode) | (3) |
Our recent studies demonstrated that the TiO2 modified with both Pt and fluoride (F-TiO2/Pt) exhibited a dual-functional photocatalytic activity for the simultaneous production of H2 and degradation of 4-CP.19,20 In Fig. 3, the H2 production on Cr2O3/Rh/SrTiO3 was compared with F-TiO2/Pt under neutral and acidic pH conditions. The photocatalytic activity of Cr2O3/Rh/SrTiO3 is higher than that of F-TiO2/Pt and less affected by the pH change. It should be noted that the activity of F-TiO2/Pt is markedly reduced at neutral pH whereas that of Cr2O3/Rh/SrTiO3 was little influenced by pH. As a result, Cr2O3/Rh/SrTiO3 is a better dual-functional photocatalyst from a practical point of view. In terms of the charge transfer characteristics, the following two major features make Cr2O3/Rh/SrTiO3 a practical dual-functional photocatalyst. For the electron transfer part, CB electrons are selectively consumed by protons only and their transfer to O2 and other electron acceptors (EA) is hindered because the Cr2O3 barrier layer is selectively permeable only to protons. On the other hand, VB holes are utilized to oxidize both H2O (to O2) and 4-CP (organic pollutants) simultaneously and the in situ generated O2 is immediately consumed for the mineralization of the organic pollutants.19,24,28 The reaction mechanisms described above are schematically illustrated in Scheme 1. In the absence of the Cr2O3 layer, the CB electrons can be consumed by not only protons but also in situ generated O2 and other reaction intermediates, which would reduce the overall dual-photocatalysis activity.
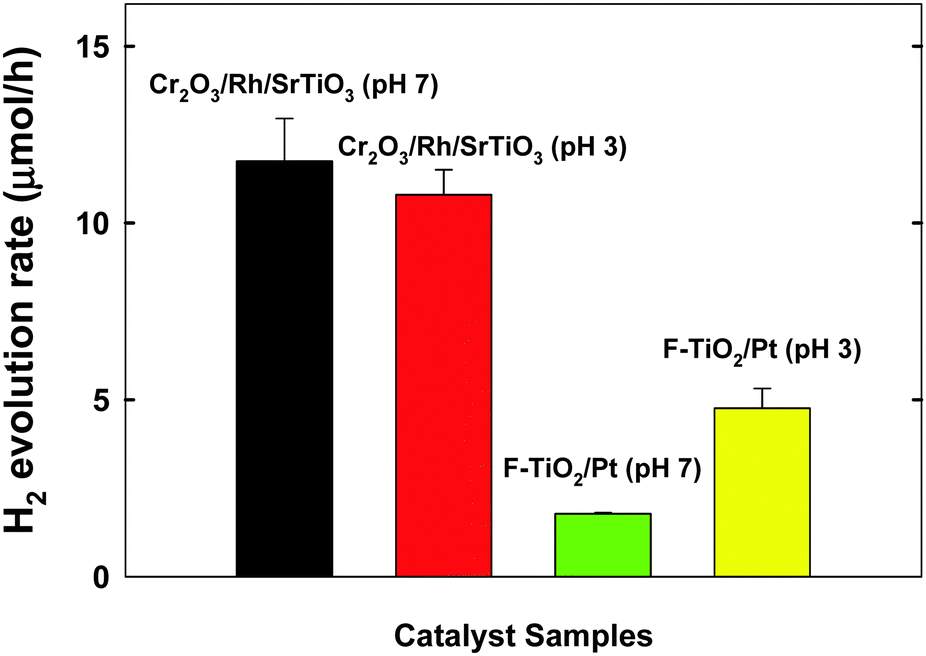 | ||
| Fig. 3 Comparison of the initial photocatalytic H2 production rate between Cr2O3/Rh/SrTiO3 and F-TiO2/Pt photocatalytic systems in the presence of 4-CP (300 μM). Experimental conditions: [catalyst] = 0.5 g L−1, λ > 320 nm, air-tight and initially Ar-purged for 1 h before UV irradiation. | ||
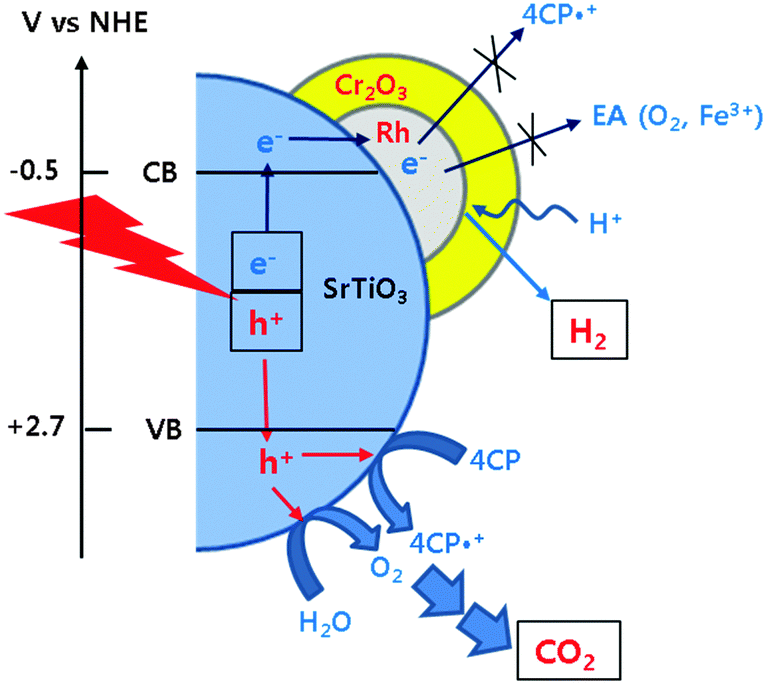 | ||
| Scheme 1 Schematic illustrations of photocatalytic reaction mechanisms occurring on the surface of Cr2O3/Rh/SrTiO3. | ||
To check the photostability of Cr2O3/Rh/SrTiO3, the photocatalytic H2 production was repeated up to four cycles in the same batch of catalyst by injecting 100 μM 4-CP every 2 h (Fig. S6, ESI†). The activity was not maintained and gradually decreased with repeated uses. To elucidate whether the gradual loss of activity was caused by the instability of the photocatalyst, the same experiment was performed without 4-CP injection. In this case, the photocatalytic activity was maintained without showing activity loss. The composite photocatalyst itself seems to be stable. Therefore, the gradual loss of photocatalytic activity observed in the presence of 4-CP might be ascribed to the accumulation of organic degradation intermediates on the catalyst surface.29,30 To further investigate the effect of organic compounds in this dual functional photocatalysis, the evolution of H2 and O2 was simultaneously measured with repeated photocatalysis cycles. In this case, 100 μM 4-CP was initially added but not replenished in the subsequent cycles. Fig. S7 (ESI†) shows that H2 production was higher in the first cycle than in the subsequent cycles: the difference in H2 production should be ascribed to the organic electron donor (i.e., 4-CP) effect. The extra holes scavenged by 4-CP make an equal number of electrons to be used for H2 production. At the same time, O2 evolved in the first cycle is immediately consumed for the mineralization of 4-CP. As a result, the ratio of H2 to O2 in the first cycle was significantly higher (H2/O2(r) = 6.9) than the stoichiometric water splitting ratio (r = 2.0). The ratio progressively approached the stoichiometric ratio as the cycle was repeated (r: 6.9 → 2.7 → 2.5 → 2.2).
In conclusion, this study demonstrated that the mineralization of organic pollutants can be achieved under the de-aerated conditions with the simultaneous H2 production over a Cr2O3/Rh/SrTiO3 photocatalyst. The present study showed the highest H2 evolution efficiency in dual-purpose photocatalysis to our knowledge. It is proposed that the Cr2O3 shell on the Rh nanoparticle core markedly enhances the H2 production and TOC removal of aromatic pollutants, which makes Cr2O3/Rh/SrTiO3 an active dual-functional photocatalyst.
This work was supported by the Global Research Laboratory (GRL) Program (NRF-2014K1A1A2041044), the Global Frontier R&D Program on Center for Multiscale Energy System (2011-0031571), and KCAP (Sogang Univ.) (No. 2009-0093880) funded by the Korea government (MSIP) through the National Research Foundation of Korea (NRF). K. M. acknowledges a Grant-in-Aid for Young Scientists (A) (Project 25709078) and the PRESTO/JST program “Chemical Conversion of Light Energy” for funding support.
References
- G. Zhang, C. Ni, X. Huang, A. Welgamage, L. A. Lawton, P. K. J. Robertson and J. T. S. Irvine, Chem. Commun., 2016, 52, 1673 RSC.
- M. Ni, D. Y. C. Leung and M. K. H. Leung, Int. J. Hydrogen Energy, 2007, 32, 3238–3247 CrossRef CAS.
- F. Sastre, M. Oteri, A. Corma and H. Garcia, Energy Environ. Sci., 2013, 6, 2211 CAS.
- Y. Tachibana, L. Vayssieres and J. R. Durrant, Nat. Photonics, 2012, 6, 511 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC.
- Y. Ma, X. Wang, Y. Jia, X. Chen, H. Han and C. Li, Chem. Rev., 2014, 114, 9987 CrossRef CAS PubMed.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520 RSC.
- H. Park, Y. Park, W. Kim and W. Choi, J. Photochem. Photobiol., C, 2013, 15, 1 CrossRef CAS.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69 CrossRef CAS.
- Q. Xiang, J. Yu and M. Jaroniec, Chem. Soc. Rev., 2012, 41, 782 RSC.
- J. Yang, R. Hu, W. Meng and Y. Du, Chem. Commun., 2016, 52, 2620 RSC.
- H. Park, H.-i. Kim, G.-h. Moon and W. Choi, Energy Environ. Sci., 2016, 9, 411 CAS.
- X.-Y. Zhang, H.-P. Li, X.-L. Cui and Y. Lin, J. Mater. Chem., 2010, 20, 2801 RSC.
- M.-C. Wu, J. Hiltunen, A. Sápi, A. Avila, W. Larsson, H.-C. Liao, M. Huuhtanen, G. Tóth, A. Shchukarev, N. Laufer, Á. Kukovecz, Z. Kónya, J.-P. Mikkola, R. Keiski, W.-F. Su, Y.-F. Chen, H. Jantunen, P. M. Ajayan, R. Vajtai and K. Kordás, ACS Nano, 2011, 5, 5025 CrossRef CAS PubMed.
- P. Gomathisankar, K. Hachisuka, H. Katsumata, T. Suzuki, K. Funasaka and S. Kaneco, Int. J. Hydrogen Energy, 2013, 38, 11840 CrossRef CAS.
- W. Zhang, Y. Wang, Z. Wang, Z. Zhong and R. Xu, Chem. Commun., 2010, 46, 7631 RSC.
- M. Wen, Y. Kuwahara, K. Mori, D. Zhang, H. Li and H. Yamashita, J. Mater. Chem. A, 2015, 3, 14134 CAS.
- Y.-J. Cho, H.-i. Kim, S. Lee and W. Choi, J. Catal., 2015, 330, 387 CrossRef CAS.
- J. Kim and W. Choi, Energy Environ. Sci., 2010, 3, 1042 CAS.
- J. Kim, D. Monllor-Satoca and W. Choi, Energy Environ. Sci., 2012, 5, 7647 CAS.
- K. Maeda, A. Xiong, T. Yoshinaga, T. Ikeda, N. Sakamoto, T. Hisatomi, M. Takashima, D. Lu, M. Kanehara, T. Setoyama, T. Teranishi and K. Domen, Angew. Chem., Int. Ed., 2010, 49, 4096 CrossRef CAS PubMed.
- K. Maeda, K. Teramura, D. Lu, N. Saito, Y. Inoue and K. Domen, Angew. Chem., Int. Ed., 2006, 45, 7806 CrossRef CAS PubMed.
- M. Yoshida, K. Takanabe, K. Maeda, A. Ishikawa, J. Kubota, Y. Sakata, Y. Ikezawa and K. Domen, J. Phys. Chem. C, 2009, 113, 10151 CAS.
- D. Hufschmidt, D. Bahnemann, J. J. Testa, C. A. Emilio and M. I. Litter, J. Photochem. Photobiol., A, 2002, 148, 223 CrossRef CAS.
- J. Kim, J. Lee and W. Choi, Chem. Commun., 2008, 756 RSC.
- C. Minero, G. Mariella, V. Maurino, D. Vione and E. Pelizzetti, Langmuir, 2000, 16, 8964 CrossRef CAS.
- H. Park and W. Choi, J. Phys. Chem. B, 2003, 107, 3885 CrossRef CAS.
- J. Theurich, M. Lindner and D. W. Bahnemann, Langmuir, 1996, 12, 6368 CrossRef CAS.
- M. I. Franch, J. Peral, X. Domenech and J. A. Ayllon, Chem. Commun., 2005, 1851 RSC.
- S. Weon and W. Choi, Environ. Sci. Technol., 2016, 50, 2556 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details of the synthesis of Rh/SrTiO3, Cr2O3/Rh/SrTiO3 and F-TiO2/Pt, experimental details of photocatalytic reaction and photoelectrochemical tests, and Fig. S1–S7. See DOI: 10.1039/c6cc04260k |
| This journal is © The Royal Society of Chemistry 2016 |