
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDetermining carbon–carbon connectivities in natural abundance organic powders using dipolar couplings†
Myriam
Dekhil
a,
Giulia
Mollica
a,
Tristan Texier
Bonniot
a,
Fabio
Ziarelli
b,
Pierre
Thureau
*a and
Stéphane
Viel
 ac
ac
aAix-Marseille Université, CNRS, ICR (UMR 7273), 13397 Marseille, France. E-mail: pierre.thureau@univ-amu.fr; Tel: +33 (0)491 288 578
bAix-Marseille Université, Centrale Marseille, CNRS, Fédération des Sciences Chimiques de Marseille (FR 1739), 13397 Marseille, France
cInstitut Universitaire de France, 75005 Paris, France
First published on 13th June 2016
Abstract
We present a solid-state NMR methodology capable of investigating the carbon skeleton of natural abundance organic powders. The methodology is based on the 13C–13C dipolar coupling interaction and allows carbon–carbon connectivities to be unambiguously established for a wide range of organic solids. This methodology is particularly suitable for disordered solids, such as natural or synthetic macromolecules, which cannot be studied using conventional diffraction or NMR techniques.
Determining the connectivity between carbon atoms is the first and most important step for the structural investigation of organic molecules. In the case of organic powders that cannot be studied by conventional diffraction techniques, solid-state nuclear magnetic resonance (SSNMR) is usually regarded as a key technique.1–6 Notably, the carbon skeleton of organic powders is usually investigated using two-dimensional correlation experiments based on 13C–13C scalar (J) couplings to transfer magnetisation between 13C nuclei separated by a single covalent bond. Although such J-based experiments have been shown to produce correlations between 13C that are not directly bonded (thereby potentially leading to wrong assignments),7 these experiments usually lead to the accurate determination of carbon connectivities and are particularly valuable to investigate samples isotopically enriched in 13C nuclei.8–18
In the case of natural-abundance (NA) samples, however, the applicability of J-based experiments is often limited. In fact, because the magnetisation transfer arising from 13C–13C J-couplings is slow (with respect to the decay of the 13C transverse magnetisation), it is typically barely detectable for NA samples. As a result, establishing carbon–carbon connectivities in NA samples using J-based experiments usually requires extensively long experimental times in the case of crystalline or semi-crystalline samples19–28 (approximately a week) and is seldom practicable in the case of NA disordered solids that display broad 13C NMR linewidths arising from fast 13C transverse magnetisation decay.29,30
Herein, we demonstrate that an unambiguous assignment of carbon connectivities in NA organic powders, especially disordered solids, can be obtained from 2D correlation experiments that are based on 13C–13C dipolar couplings.30,31 An important feature of dipolar-based experiments is that the magnetisation transfer builds up much faster than in J-based experiments, and hence dipolar-based experiments are suitable for organic solids with fast 13C transverse magnetisation decays. Moreover, we show here that a magnetisation transfer time of 0.4 ms leads exclusively to correlation signals between covalently bonded 13C–13C pairs. In contrast, signals arising from 13C–13C pairs separated by more than one covalent bond are at least 5 times weaker, and thus would not be detected in 2D 13C–13C DQ dipolar correlation spectra of NA samples (ESI†).
Fig. 1 shows the pulse sequence for magic-angle spinning (MAS) 2D dipolar-based 13C–13C correlation experiments. The recoupling of the 13C–13C dipolar interaction is based on the R2092 symmetry,32–34 which generates during the first delay τDQ double quantum (DQ) coherences that subsequently evolve during t1 at the sum of the chemical shift frequencies of pairs of coupled 13C–13C nuclei, before being reconverted during the second τDQ into single quantum coherences for detection during t2. It should be noted that, for NA samples, DQ coherences are useful not only for straightforward data interpretation but also to suppress intense signals arising from the large number of uncoupled 13C nuclei.
 | ||
| Fig. 1 Pulse sequence for 2D 13C–13C correlation experiments based on dipolar coupling (a). The RNνn symmetry was applied during the delays τDQ to excite and reconvert double quantum (DQ) coherences, yielding correlation peaks in the indirect dimension of the 2D spectrum at the sum of the chemical shifts of the 2 coupled 13C nuclei. The selected symmetry (N = 20, n = 2 and ν = 9) led to 10 Rϕ,R−ϕ′ elements fitting into 2 rotor periods, with ϕ = 81° (b). The basic element is shown in (c) and it consists of a composite 180° pulse. | ||
Interestingly, the R2092 symmetry is particularly robust with respect to 1H–13C dipolar coupling and 13C chemical shift anisotropy (CSA).32,35–37 Notably, no 1H decoupling is required during the 13C recoupling periods τDQ32–34 and, thus, this experiment is very easy to implement (only the 13C 90° pulse needs to be calibrated). It should be noted that the R2092 symmetry is appropriate for a MAS frequency of 10 kHz. In this case, a 4 mm MAS rotor can be used, providing the best compromise between MAS frequency and sensitivity for NA samples at 9.4 T.
Furthermore, using our instrumentation, the DQ recoupling of the 13C–13C dipolar interaction using the R2092 symmetry is 1.7 times more efficient than standard dipolar recoupling symmetries, such as POST-C738 or SPC539 (see the ESI†). This enhancement translates in substantial shorter experimental times, i.e. 2.9 times faster, and as a result, 2D 13C–13C correlation experiments for NA samples can be obtained within practicable experimental durations using standard solid-state NMR hardware at room temperature.
The 2D dipolar-based 13C–13C correlation spectrum for a powder sample of L-tyrosine ethyl ester is shown in Fig. 2. The 2D spectrum was recorded using a 9.4 T magnet and a MAS frequency of 10 kHz; the experimental time was 3 days. The 1D 13C cross-polarisation (CP) MAS spectrum is also shown above the 2D spectrum. Remarkably, the resonances corresponding to aromatic carbons are significantly broader than the other 13C signals. This broadening results from the fast 13C transverse magnetisation decay arising from molecular motion.40,41 As a result, correlation signals involving the aromatic carbons are not detectable using J-based correlation experiments.22 In contrast, all the 13C–13C correlation signals between covalently bonded 13C nuclei are observed in the 2D dipolar-based 13C–13C correlation spectrum with excellent resolution and sensitivity.
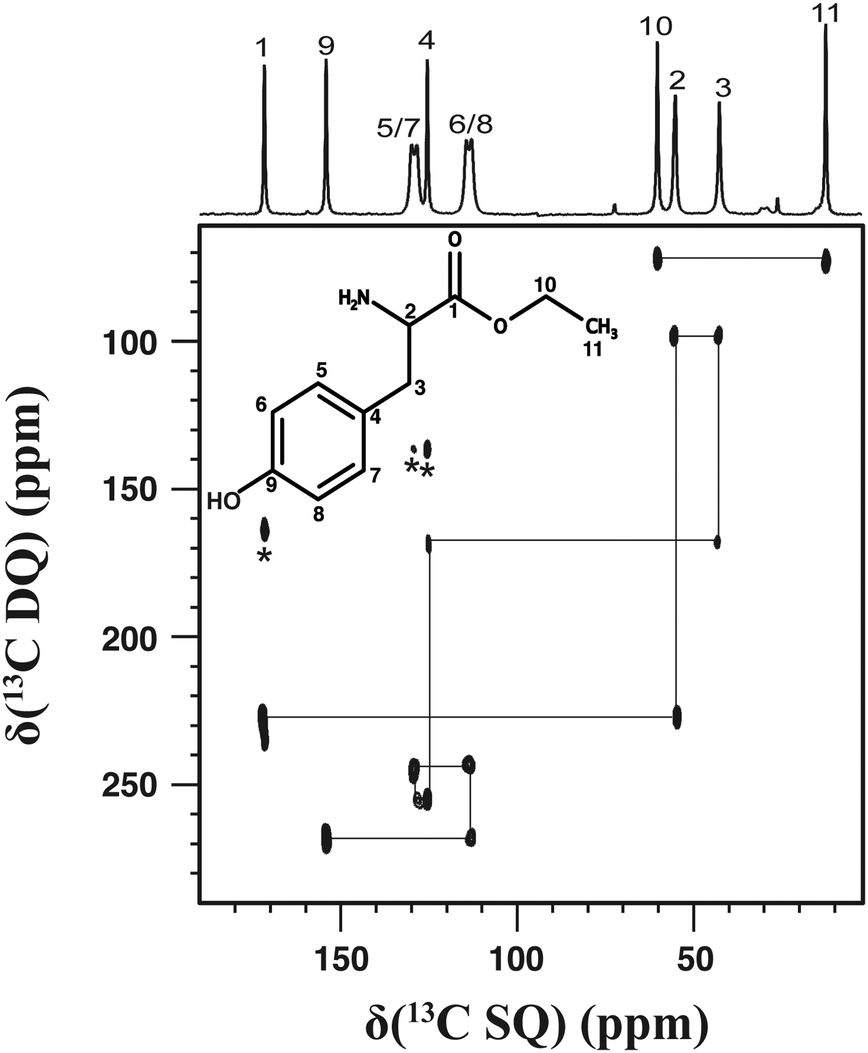 | ||
| Fig. 2 13C–13C DQ dipolar correlation spectrum of L-tyrosine ethyl ester powder. The spectrum was recorded using τDQ = 0.4 ms, a MAS frequency of 10 kHz, 27 t1 increments and 1600 repetitions for each t1 increment. The repetition delay was 3 s. The total experimental time was 3 days. The 13C CPMAS spectrum is shown in the top projection. The symbol (*) indicates spinning sidebands. | ||
Fig. 3 illustrates the capabilities of 2D dipolar-based 13C–13C correlation experiments to investigate a disordered solid, here, an amorphous poly(methyl methacrylate) (PMMA) sample. This spectrum has been recorded in less than 2 days. Interestingly, the 13C signals of PMMA arise from both mobile and rigid 13C sites. For example, the OCH3 or CO resonances correspond to the mobile part of PMMA while the CH2 or CH3 resonances correspond to the more rigid backbone chain.42 We notice in Fig. 3 that, notwithstanding significant mobility differences, all the correlation peaks arising from covalently bonded 13C–13C pairs could be observed in the 2D 13C–13C DQ dipolar correlation spectrum.‡ We emphasise that 2D dipolar-based 13C–13C correlation experiments are suitable to samples that give rise to observable signals in 13C CPMAS experiments. In the case of samples with high molecular mobility, which are intrinsically unsuitable for 13C CPMAS experiments,43 it would be more advantageous to directly excite 13C nuclei and to explore 13C–13C connectivities through J-based experiments.30 Nevertheless, we have shown here (Fig. 2 and 3) that 2D dipolar-based 13C–13C correlation experiments are useful even for solids subjected to molecular motions.
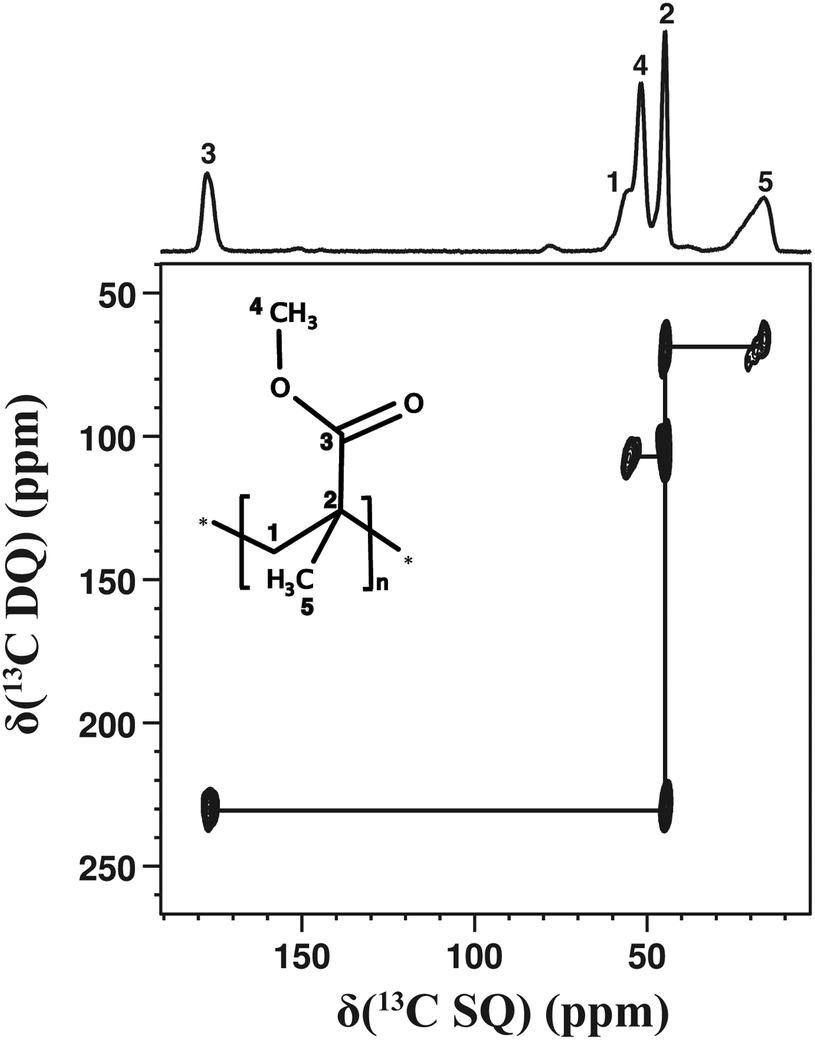 | ||
| Fig. 3 13C–13C DQ dipolar correlation spectrum of a synthetic polymer, the poly(methyl methacrylate) (PMMA), recorded with τDQ = 0.4 ms, a MAS frequency of 10 kHz, 38 t1 increments and 1024 repetitions for each t1 increment. The repetition delay was set to 2 s. The total experimental time was 1.8 days. The 13C CPMAS spectrum is shown in the top projection. | ||
An additional example is shown in Fig. 4 for cellulose, a natural polymer containing both amorphous and crystalline phases. The 1D 13C CPMAS spectrum is shown above the 2D 13C–13C DQ dipolar correlation spectrum. In the 1D 13C CPMAS spectrum the apparent line widths range from 400 to 600 Hz and are partly related to the disordered character of the sample. The 2D 13C–13C DQ dipolar correlation spectrum was recorded within 3 days and all one-bond correlations were observed with very good sensitivity and satisfactory resolution. Notably, the assignment based on dipolar coupling experiments is in agreement with the assignment based on J-coupling experiments.§![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 29 Moreover, correlation peaks corresponding to both amorphous and crystalline signals are observed in the 2D 13C–13C DQ dipolar correlation spectrum, as exemplified for the signals at 86 ppm and 63 ppm.
29 Moreover, correlation peaks corresponding to both amorphous and crystalline signals are observed in the 2D 13C–13C DQ dipolar correlation spectrum, as exemplified for the signals at 86 ppm and 63 ppm.
 | ||
| Fig. 4 13C–13C DQ dipolar correlation spectrum of cellulose, recorded with τDQ = 0.4 ms, a MAS frequency of 10 kHz, 20 t1 increments and 2048 repetitions for each t1 increment. The repetition delay was 3.2 s. The total experimental time was approximately 3 days. The 13C CPMAS spectrum is shown in the top projection. | ||
In summary, we have shown that unambiguous carbon–carbon connectivities in NA organic solids can be determined using dipolar-based 13C–13C correlation experiments. These experiments can be performed within practicable experimental times and are suitable for different kinds of samples, such as molecular crystals and disordered solids including synthetic or natural polymers. This methodology is complementary to J-based 13C–13C correlation experiments, especially in the case of NA disordered solids having fast 13C transverse magnetisation decay. Finally, the methodology presented here can be carried out at room temperature and is thus complementary to 13C–13C correlation experiments enhanced with dynamic nuclear polarisation,44–47 which require samples that can be analysed at cryogenic temperatures and in the presence of paramagnetic dopants.
This work has been carried out thanks to the support from the French National Research Agency (ANR) grant number ANR-13-JS080001.
References
- D. Massiot, R. J. Messinger, S. Cadars, M. Deschamps, V. Montouillout, N. Pellerin, E. Veron, M. Allix, P. Florian and F. Fayon, Acc. Chem. Res., 2013, 46, 1975–1984 CrossRef CAS PubMed
.
- R. J. Messinger, K. Na, Y. Seo, R. Ryoo and B. F. Chmelka, Angew. Chem., Int. Ed., 2015, 54, 927–931 CrossRef CAS PubMed
- M. R. Hansen, R. Graf and H. W. Spiess, Chem. Rev., 2016, 116, 1272–1308 CrossRef CAS PubMed
- S. Radhakrishnan, P.-J. Goossens, P. C. M. M. Magusin, S. P. Sree, C. Detavernier, E. Breynaert, C. Martineau, F. Taulelle and J. A. Martens, J. Am. Chem. Soc., 2016, 138, 2802–2808 CrossRef CAS PubMed
- C. M. Widdifield, H. Robson and P. Hodgkinson, Chem. Commun., 2016, 52, 6685–6688 RSC
- S. E. Ashbrook and D. McKay, Chem. Commun., 2016, 52, 7186–7204 RSC
- F. Fayon, D. Massiot, M. H. Levitt, J. J. Titman, D. H. Gregory, L. Duma, L. Emsley and S. P. Brown, J. Chem. Phys., 2005, 122, 194313 CrossRef PubMed
- G. Grasso, T. M. de Swiet and J. J. Titman, J. Phys. Chem. B, 2002, 106, 8676–8680 CrossRef CAS
- G. De Paepe, N. Giraud, A. Lesage, P. Hodgkinson, A. Böckmann and L. Emsley, J. Am. Chem. Soc., 2003, 125, 13938–13939 CrossRef CAS PubMed
- E. H. Hardy, A. Detken and B. H. Meier, J. Magn. Reson., 2003, 165, 208–218 CrossRef CAS PubMed
- S. Cadars, A. Lesage and L. Emsley, J. Am. Chem. Soc., 2005, 127, 4466–4476 CrossRef CAS PubMed
- L. Chen, R. A. Olsen, D. W. Elliott, J. M. Boettcher, D. H. Zhou, C. M. Rienstra and L. J. Mueller, J. Am. Chem. Soc., 2006, 128, 9992–9993 CrossRef CAS PubMed
- F. Fayon, C. Roiland, L. Emsley and D. Massiot, J. Magn. Reson., 2006, 179, 49–57 CrossRef CAS PubMed
- R. Witter, U. Sternberg, S. Hesse, T. Kondo, F.-T. Koch and A. S. Ulrich, Macromolecules, 2006, 39, 6125–6132 CrossRef CAS
- L. Chen, J. M. Kaiser, T. Polenova, J. Yang, C. M. Rienstra and L. J. Mueller, J. Am. Chem. Soc., 2007, 129, 10650–10651 CrossRef CAS PubMed
- N. Baccile, G. Laurent, F. Babonneau, F. Fayon, M.-M. Titirici and M. Antonietti, J. Phys. Chem. C, 2009, 113, 9644–9654 CrossRef CAS
- D. Lee, J. Struppe, D. W. Elliott, L. J. Mueller and J. J. Titman, Phys. Chem. Chem. Phys., 2009, 11, 3547–3553 RSC
- X. Liu, W. Chen, Y. lee Hong, S. Yuan, S. Kuroki and T. Miyoshi, Macromolecules, 2015, 48, 5300–5309 CrossRef CAS
- A. Lesage, C. Auger, S. Caldarelli and L. Emsley, J. Am. Chem. Soc., 1997, 119, 7867–7868 CrossRef CAS
- R. A. Olsen, J. Struppe, D. W. Elliott, R. J. Thomas and L. J. Mueller, J. Am. Chem. Soc., 2003, 125, 11784–11785 CrossRef CAS PubMed
- H. Kono, T. Erata and M. Takai, Macromolecules, 2003, 36, 5131–5138 CrossRef CAS
- G. De Paëpe, A. Lesage, S. Steuernagel and L. Emsley, ChemPhysChem, 2004, 5, 869–875 CrossRef PubMed
- H. Kono, Biopolymers, 2004, 75, 255–263 CrossRef CAS PubMed
- J. Brus and A. Jegorov, J. Phys. Chem. A, 2004, 108, 3955–3964 CrossRef CAS
- R. K. Harris, S. A. Joyce, C. J. Pickard, S. Cadars and L. Emsley, Phys. Chem. Chem. Phys., 2006, 8, 137–143 RSC
- R. K. Harris, S. Cadars, L. Emsley, J. R. Yates, C. J. Pickard, R. K. R. Jetti and U. J. Griesser, Phys. Chem. Chem. Phys., 2007, 9, 360–368 RSC
- M. Deschamps, S. Cadars, E. Gilbert, P. Azas, E. Raymundo-Pinero, F. Béguin and D. Massiot, Solid State Nucl. Magn. Reson., 2012, 42, 81–86 CrossRef CAS PubMed
- M. Baias, J.-N. Dumez, P. H. Svensson, S. Schantz, G. M. Day and L. Emsley, J. Am. Chem. Soc., 2013, 135, 17501–17507 CrossRef CAS PubMed
- A. Lesage, M. Bardet and L. Emsley, J. Am. Chem. Soc., 1999, 121, 10987–10993 CrossRef CAS
- Y. Su and M. Hong, J. Phys. Chem. B, 2011, 115, 10758–10767 CrossRef CAS PubMed
- M. Hong, J. Magn. Reson., 1999, 136, 86–91 CrossRef CAS PubMed
- M. Carravetta, M. Eden, O. G. Johannessen, H. Luthman, P. J. E. Verdegem, J. Lugtenburg, A. Sebald and M. H. Levitt, J. Am. Chem. Soc., 2001, 123, 10628–10638 CrossRef CAS PubMed
-
M. H. Levitt, in Encyclopedia of Nuclear Magnetic Resonance, ed. D. M. Grant and R. Harris, John Wiley & Sons, Ltd, Chichester, 2002, vol. 9, p. 165 Search PubMed
-
M. Eden, eMagRes, John Wiley & Sons, Ltd, 2013, vol. 2, pp. 351–364 Search PubMed
- C. E. Hughes, S. Luca and M. Baldus, Chem. Phys. Lett., 2004, 385, 435–440 CrossRef CAS
- I. Marin-Montesinos, D. H. Brouwer, G. Antonioli, W. C. Lai, A. Brinkmann and M. H. Levitt, J. Magn. Reson., 2005, 177, 307–317 CrossRef PubMed
- O. Lafon, J. Trebosc, B. Hu, G. De Paepe and J.-P. Amoureux, Chem. Commun., 2011, 47, 6930–6932 RSC
- M. Hohwy, H. J. Jakobsen, M. Edén, M. H. Levitt and N. C. Nielsen, J. Chem. Phys., 1998, 108, 2686 CrossRef CAS
- M. Hohwy, C. M. Rienstra, C. P. Jaroniec and R. G. Griffin, J. Chem. Phys., 1999, 110, 7983 CrossRef CAS
- L. Frydman, S. Vallabhaneni, Y. K. Lee and L. Emsley, J. Chem. Phys., 1994, 101, 111–117 CrossRef CAS
- X. Helluy and A. Sebald, J. Phys. Chem. B, 2003, 107, 3290–3296 CrossRef CAS
- E. R. deAzevedo, W.-G. Hu, T. J. Bonagamba and K. Schmidt-Rohr, J. Am. Chem. Soc., 1999, 121, 8411–8412 CrossRef CAS
- R. Zhang, K. H. Mroue and A. Ramamoorthy, J. Magn. Reson., 2016, 266, 59–66 CrossRef CAS PubMed
- A. J. Rossini, A. Zagdoun, F. Hegner, M. Schwarzwälder, D. Gajan, C. Copéret, A. Lesage and L. Emsley, J. Am. Chem. Soc., 2012, 134, 16899–16908 CrossRef CAS PubMed
- H. Takahashi, D. Lee, L. Dubois, M. Bardet, S. Hediger and G. De Paepe, Angew. Chem., Int. Ed., 2012, 51, 11766–11769 CrossRef CAS PubMed
- G. Mollica, M. Dekhil, F. Ziarelli, P. Thureau and S. Viel, Angew. Chem., Int. Ed., 2015, 54, 6028–6031 CrossRef CAS PubMed
- R. L. Johnson, F. A. Perras, T. Kobayashi, T. J. Schwartz, J. A. Dumesic, B. H. Shanks and M. Pruski, Chem. Commun., 2016, 52, 1859–1862 RSC
Footnotes |
| † Electronic supplementary information (ESI) available: Predicted signal intensities, experimental recoupling efficiencies, experimental details. See DOI: 10.1039/c6cc04202c |
| ‡ Molecular mobility could potentially reduce the DQ correlation intensity as a result of partial averaging of the 13C–13C DQ dipolar coupling. |
| § J-coupling experiments were recorded on a partially 13C enriched cellulose sample. |
| This journal is © The Royal Society of Chemistry 2016 |