
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diversity in a simple co-crystal: racemic and kryptoracemic behaviour†
U. B.
Rao Khandavilli
a,
Matteo
Lusi
 b,
Balakrishna R.
Bhogala
a,
Anita R.
Maguire
c,
Matthias
Stein
d and
Simon E.
Lawrence
*a
b,
Balakrishna R.
Bhogala
a,
Anita R.
Maguire
c,
Matthias
Stein
d and
Simon E.
Lawrence
*a
aDepartment of Chemistry, Analytical and Biological Chemistry Research Facility, Synthesis and Solid State Pharmaceutical Centre, University College Cork, Ireland. E-mail: simon.lawrence@ucc.ie
bDepartment of Chemical & Environmental Sciences, University of Limerick, Limerick, Ireland
cDepartment of Chemistry and School of Pharmacy, Analytical and Biological Chemistry Research Facility, Synthesis and Solid State Pharmaceutical Centre, University College Cork, Cork, Ireland
dMolecular Simulations and Design Group, Max Planck Institute for Dynamics of Complex Technical Systems, Magdeburg, Germany
First published on 8th June 2016
Abstract
The crystal structure containing (±)-3-methyl-2-phenylbutyramide with salicylic acid is the first example of a kryptoracemate co-crystal. It exhibits the first temperature mediated reversible single-crystal to single-crystal transition between two kryptoracemate forms, in addition to crystallising in another, racemic, form. Theoretical calculations and structural analysis reveal that there are only small differences in both energy and packing arrangements between the three forms. These results suggest that co-crystals can be an opportunity to investigate kryptoracemate behaviour.
Understanding the solid state behaviour of organic compounds has become increasingly important in a variety of fields over the past three decades.1–4 Co-crystallization is one approach that has garnered interest since the generation of families of related materials provides an opportunity to modulate targeted physicochemical properties over a broad range. For this reason it has been explored in a variety of fields including pharmaceuticals,5–9 nutraceuticals,10–12 agrochemicals,13,14 explosives,15–18 solid-state photochemistry,19,20 chiral resolution,21–23etc. Recently, the US Food and Drug Administration recognised the importance of co-crystals and issued guidelines for their use in the pharmaceutical sector.24 Separation of enantiomers has developed in recent years, particularly with the advent of Viedma ripening.25–27 Enantiomer separation using co-crystallization is less well investigated in comparison to salt formation,28,29 even though it is, in theory, more applicable since it does not require the presence of an ionisable functional group in the molecule of interest.2 With this background, we have chosen to investigate the co-crystallisation behaviour of (±)-3-methyl-2-phenylbutyramide, 1, an anti-mitotic compound.30
In our co-crystallisation screening of 1 we obtained co-crystals with salicylic acid, 1·SA. Structural analysis revealed the co-crystal crystallised in a Sohncke (chiral) space group with both enantiomers present in the asymmetric unit. Racemic compounds which crystallise in chiral space groups yet remain racemic (i.e. maintain a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of enantiomers) are rare, with limited discussion found in the literature.31–37 Morales & Fronczek coined the term “kryptoracemate” for these systems in 1996.31 Fábián and Brock examined their occurrence amongst organic molecules by analysing the Cambridge Structural Database (CSD) in 2010; finding only 151 organic kryptoracemate structures at that time.34 Very recently, Laubenstein et al. examined the first example of a molecule crystallising in both racemic and kryptoracemic structures,36 and a second example has just been published.37 To the best of our knowledge the co-crystal of 1 with salicylic acid is the first co-crystal kryptoracemate. Further work uncovered a racemic form, plus the first example of a single-crystal to single-crystal transformation between two kryptoracemic forms.
:1 ratio of enantiomers) are rare, with limited discussion found in the literature.31–37 Morales & Fronczek coined the term “kryptoracemate” for these systems in 1996.31 Fábián and Brock examined their occurrence amongst organic molecules by analysing the Cambridge Structural Database (CSD) in 2010; finding only 151 organic kryptoracemate structures at that time.34 Very recently, Laubenstein et al. examined the first example of a molecule crystallising in both racemic and kryptoracemic structures,36 and a second example has just been published.37 To the best of our knowledge the co-crystal of 1 with salicylic acid is the first co-crystal kryptoracemate. Further work uncovered a racemic form, plus the first example of a single-crystal to single-crystal transformation between two kryptoracemic forms.
Initially, 1·SA was isolated from dichloromethane at room temperature. Single crystal X-ray diffraction reveals that at room temperature the crystals are in the orthorhombic space group P212121; Form I. Upon cooling to 100 K on the diffractometer, Form I transformed to the lower symmetry monoclinic Sohncke space group P21 (Form II). Variable temperature single crystal X-ray diffraction confirmed that the transformation between the two forms is reversible, occurring between 150 K and 250 K (ESI†).
Crystallisation screening using various solvents and solvent mixtures by crystallization from a solution at room temperature failed to produce Form II, although in some cases a novel racemic polymorph of 1·SA (Form III) was observed concomitantly with Form I (ESI†). This new form crystallises in the centrosymmetric space group P21/c. Pure Form III was obtained from dichloromethane at 0 °C. Thus, it was possible to obtain pure samples of Forms I and III for further analysis, whereas Form II could only be obtained upon cooling of Form I, as summarised in Scheme 1.
 | ||
| Scheme 1 Schematic representation of the co-crystallization outcomes. | ||
The three polymorphs were further investigated by spectroscopic techniques. The IR and Raman spectra of Forms I and III are almost identical, with only very slight variations observed in the lattice vibrational region (ESI†). Powder X-ray diffraction, PXRD, data for the bulk material is consistent with the single crystal data for both Form I and III (ESI†), highlighting that the structural analyses obtained from the single crystal experiments are representative of the bulk material. DSC thermograms between RT and 150 °C indicate no interconversion between these two forms and show that Form I is the lower melting polymorph (85–88 °C for Form I vs. 86–89 °C for Form III). Melting of Form III requires higher energy (3.4 vs. 4.7 W g−1) suggesting that it is the most stable polymorph (ESI†).
All three forms have similar unit cell axes and volumes, differing only in one unit cell angle (ESI†). In Form I there are two symmetry independent salicylic acid molecules along with two molecules of 1 (designated R1 & S2) and since it is a non-centrosymmetric structure there are no corresponding S1 and R2 molecules present. In Form II, four symmetry independent salicylic acid molecules and corresponding amide molecules (R1, S2, R3 and S4) are present. Form III has two symmetry independent salicylic acid molecules and two amide molecules (R1 and S2) and, being centrosymmetric, S1 and R2 are also present.
The molecular conformations in the three forms are very similar, the main differences being slight variations in the torsion angles between the phenyl group and the chiral centre of 1 (Fig. 1 and ESI†). In each case, the hydroxyl group of the salicylic acid is involved in intramolecular O–H⋯O hydrogen bonding [S(6) using Etter's notation38,39] and is not involved in the formation of the supramolecular architecture. This behaviour is common for salicylic acid and other ortho-substituted hydroxyl carboxylic acids.40–42 The three forms all exhibit hydrogen bonding from a salicylic acid molecule to one molecule of 1 (either R or S) via the well-known 8-membered acid-amide R22(8) heterosython and one-dimensional tapes via C(4) N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C hydrogen bonding catamers, using the anti N–H hydrogen atom (Fig. 1). The tapes alternate between R and S enantiomers of 1, Fig. 2. Differences between the three forms are due to the spatial arrangement of these tapes. In Form I, tape A is related to tapes B, C and D by a 21 screw axis along the x, z and y axes, respectively. The tapes A′ and B′ are in the adjacent unit cell to A and B. Form II has a very similar spatial arrangement to Form I. The tapes are slipped relative to one another along the monoclinic direction causing the loss of symmetry and the shortening of the π–π interactions between the salicylic acid from about 3.9 Å (Form I) to 3.6 Å (Form II), Fig. 3. Loss of the two 21 screw symmetries along the a and c axes means there is an increase in the number of symmetry independent molecules present in the lattice (from Z′ = 2 to 4). In Form III the tapes are arranged in anti-parallel fashion along both the b and c axes over crystallographic inversion centres.
C hydrogen bonding catamers, using the anti N–H hydrogen atom (Fig. 1). The tapes alternate between R and S enantiomers of 1, Fig. 2. Differences between the three forms are due to the spatial arrangement of these tapes. In Form I, tape A is related to tapes B, C and D by a 21 screw axis along the x, z and y axes, respectively. The tapes A′ and B′ are in the adjacent unit cell to A and B. Form II has a very similar spatial arrangement to Form I. The tapes are slipped relative to one another along the monoclinic direction causing the loss of symmetry and the shortening of the π–π interactions between the salicylic acid from about 3.9 Å (Form I) to 3.6 Å (Form II), Fig. 3. Loss of the two 21 screw symmetries along the a and c axes means there is an increase in the number of symmetry independent molecules present in the lattice (from Z′ = 2 to 4). In Form III the tapes are arranged in anti-parallel fashion along both the b and c axes over crystallographic inversion centres.
 | ||
| Fig. 1 The hydrogen bonding in Form 1 showing the 1-D chain and acid-amide dimers, left, and overlay of all three forms (red = Form I, violet = Form II and yellow = Form III), right. | ||
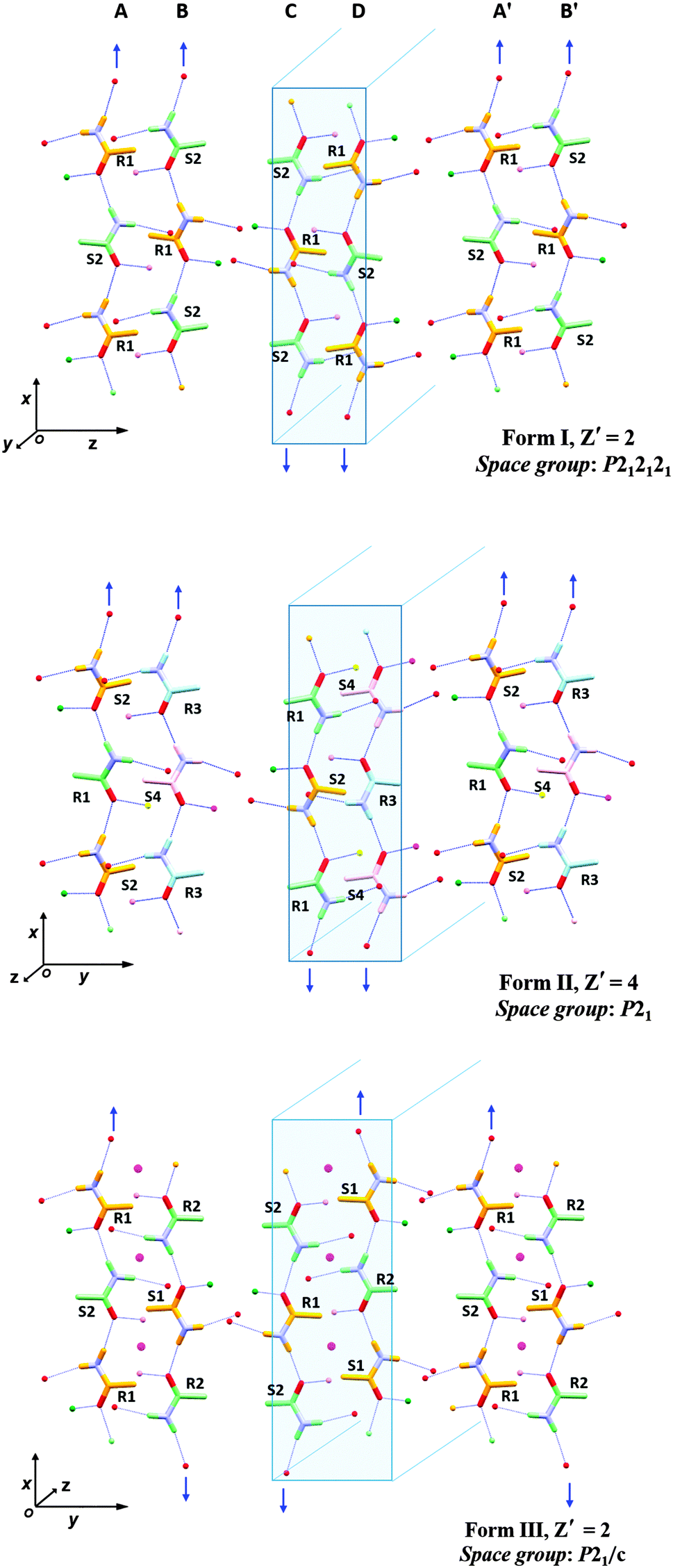 | ||
| Fig. 2 Packing diagrams of Form I–III highlighting the parallel (Forms I and II) and anti-parallel (Form III) arrangement of the N–H⋯OC C(4) hydrogen bonded catamers (blue arrows indicate H⋯O direction, blue box for clarity of catamers). Symmetry related molecules have the same colour for the C and H atoms and the molecules are labelled with their chirality. Only the amide functional group of 1 and H-bond connectivity are shown for clarity. Magenta dots indicate the inversion centre in Form III. | ||
 | ||
| Fig. 3 π–π interactions in Form I (left) and Form II (right) viewed perpendicular to the ab and bc plane respectively. The red arrow shows the relative slippage of the molecules. | ||
In general, solid state transitions between polymorphs occur through reconstructive, topotactic or epitactic mechanisms and are usually activated by heat, light or mechanical stress.43–47 Often this results in significant loss of crystallinity due to the restricted molecular mobility in crystals and, hence, single-crystal to single-crystal transitions are relatively rare in organic compounds. The similar crystal packing in Forms I and II enables the rationalisation of the single-crystal to single-crystal reversible transformation. Solid-state transformation between Forms I and III was not observed; most likely because the inversion centre in Form III requires a reconstructive pathway for interconversion between the parallel (Form I) and anti-parallel (Form III) packing arrangements.
High Z′ values have been discussed in an extensive review by Steed and Steed.4 In this context 1·SA offers an opportunity to determine the energy relationship between structures with different Z′ values (Form I vs. Form II) and whether the kryptoracemates are metastable with respect to the centrosymmetric racemate (Form II vs. Form III). According to Oswald's rule of stages, metastable forms crystallize first and the thermodynamically stable forms crystallize later.48–50 In the recent study by Laubenstein et al., the kryptoracemate was initially identified and three centrosymmetric polymorphs subsequently found in a polymorphic screen.36 Therefore, we have investigated the energies of the three forms of 1·SA with periodic DFT calculations using the revPBE functional with dispersion correction. These confirm the monoracemate Form III to be the minimum structure. Forms I and II are very close in energy and interconversion between them seems to be possible without large distortions. It must be noted that the large unit cells with hundreds of atoms, hydrogen bonding interactions in planes and subtle π-interactions between them pose a challenge to current theoretical approaches.
The relative stability of Forms I and III was investigated by slurrying in HPLC-grade water for 48 hours (ESI†). There was no change observed for the experiments involving the pure forms. Form III was the only product observed for the experiments beginning with mixtures of the two forms, highlighting that Form III is the more stable form, in agreement with the computational and DSC data.
Price et al. noted the large number of known and predicted structures for organic compounds and suggested that it is unlikely that crystallization will result in the formation of only one racemic or two separate enantiopure crystal forms.51 Their work on mandelic acids showed a range of thermodynamically feasible structures of enantiopure and racemic forms with similar energies. In our work, even though these kryptoracemate co-crystals have only one chiral component, their similar lattice energies suggest that kryptoracemates may need to be considered when designing processes for chiral separation, especially as their presence is an obstacle to this overall goal.
In summary, we have reported two kryptoracemates and one racemate of a co-crystal containing (±)-3-methyl-2-phenylbutyramide with salicylic acid. The two kryptoracemates are enantiotropically related by a reversible phase transition that can be observed in single crystals. The relationship between the kryptoracemates and the racemate is difficult to determine with high accuracy both experimentally and computationally. It will be interesting to see whether kryptoracemates remain metastable forms with respect to their racemic counterparts for other systems, or are genuinely the most thermodynamically stable form.
This publication has emanated from research conducted with the financial support of Science Foundation Ireland under Grant Number 12/RC/2275 and the Max Planck Society for the Advancement of Science. The authors would like to acknowledge financial support, in part, by the COST Action CM1402, Crystallize.
Notes and references
- D. Braga, S. d'Agostino, E. Dichiarante, L. Maini and F. Grepioni, Chem. – Asian J., 2011, 6, 2214–2223 CrossRef CAS PubMed.
- N. K. Duggirala, M. L. Perry, Ö. Almarsson and M. J. Zaworotko, Chem. Commun., 2016, 52, 640–655 RSC.
- A. J. Cruz-Cabeza, S. M. Reutzel-Edens and J. Bernstein, Chem. Soc. Rev., 2015, 44, 8619–8635 RSC.
- K. M. Steed and J. W. Steed, Chem. Rev., 2015, 115, 2895–2933 CrossRef CAS PubMed.
- P. Vishweshwar, J. A. McMahon, J. A. Bis and M. J. Zaworotko, J. Pharm. Sci., 2006, 95, 499–516 CrossRef CAS PubMed.
- N. Qiao, M. Li, W. Schlindwein, N. Malek, A. Davies and G. Trappitt, Int. J. Pharm., 2011, 419, 1–11 CrossRef CAS PubMed.
- R. Thakuria, A. Delori, W. Jones, M. P. Lipert, L. Roy and N. Rodríguez-Hornedo, Int. J. Pharm., 2013, 453, 101–125 CrossRef CAS PubMed.
- H. G. Brittain, J. Pharm. Sci., 2013, 102, 311–317 CrossRef CAS PubMed.
- C. B. Aakeröy, S. Forbes and J. Desper, J. Am. Chem. Soc., 2009, 131, 17048–17049 CrossRef PubMed.
- A. S. Sinha, A. R. Maguire and S. E. Lawrence, Cryst. Growth Des., 2015, 15, 984–1009 CAS.
- A. S. Sinha, U. B. R. Khandavilli, E. L. O'Connor, B. J. Deadman, A. R. Maguire and S. E. Lawrence, CrystEngComm, 2015, 17, 4832–4841 RSC.
- N. Schultheiss, S. Bethune and J.-O. Henck, CrystEngComm, 2010, 12, 2436–2442 RSC.
- E. Nauha, Crystalline forms of selected agrochemical actives: design and synthesis of co-crystals, Department of Chemistry, University of Jyväskylä, Research report No., 151, 2012.
- E. Nauha and M. Nissinen, J. Mol. Struct., 2011, 1006, 566–569 CrossRef CAS.
- O. Bolton, L. R. Simke, P. F. Pagoria and A. J. Matzger, Cryst. Growth Des., 2012, 12, 4311–4314 CAS.
- K. B. Landenberger, O. Bolton and A. J. Matzger, Angew. Chem., Int. Ed., 2013, 52, 6468–6471 CrossRef CAS PubMed.
- J. Evers, I. Gospodinov, M. Joas, T. M. Klapötke and J. Stierstorfer, Inorg. Chem., 2014, 53, 11749–11756 CrossRef CAS PubMed.
- K. Liu, G. Zhang, J. Luan, Z. Chen, P. Su and Y. Shu, J. Mol. Struct., 2016, 1110, 91–96 CrossRef CAS.
- B. R. Bhogala, B. Captain, A. Parthasarathy and V. Ramamurthy, J. Am. Chem. Soc., 2010, 132, 13434–13442 CrossRef CAS PubMed.
- M. A. Sinnwell and L. R. MacGillivray, Angew. Chem., Int. Ed., 2016, 55, 3477–3480 CrossRef CAS PubMed.
- O. Sánchez-Guadarrama, F. Mendoza-Navarro, A. Cedillo-Cruz, H. Jung-Cook, J. I. Arenas-García, A. Delgado-Díaz, D. Herrera-Ruiz, H. Morales-Rojas and H. Höpfl, Cryst. Growth Des., 2016, 16, 307–314 Search PubMed.
- S. Iwama, K. Kuyama, Y. Mori, K. Manoj, R. G. Gonnade, K. Suzuki, C. E. Hughes, P. A. Williams, K. D. M. Harris, S. Veesler, H. Takahashi, H. Tsue and R. Tamura, Chem. – Eur. J., 2014, 20, 10343–10350 CrossRef CAS PubMed.
- G. Springuel, L. Collard and T. Leyssens, CrystEngComm, 2013, 15, 7951–7958 RSC.
- Centre for Drug Evaluation and Research, Guidance for industry: Regulatory Classification of Pharmaceutical Cocrystals, Rockville, Maryland: United States Food and Drug Administration, April, 2013.
- H. Lorenz and A. Seidel-Morgenstern, Angew. Chem., Int. Ed., 2014, 53, 1218–1250 CrossRef CAS PubMed.
- R. R. E. Steendam, J. M. M. Verkade, T. J. B. van Benthem, H. Meekes, W. J. P. van Enckevort, J. Raap, F. P. J. T. Rutjes and E. Vlieg, Nat. Commun., 2014, 5, 5543 CrossRef CAS PubMed.
- L.-C. Sogutoglu, R. R. E. Steendam, H. Meekes, E. Vlieg and F. P. J. T. Rutjes, Chem. Soc. Rev., 2015, 44, 6723–6732 RSC.
- G. Springuel and T. Leyssens, Cryst. Growth Des., 2012, 12, 3374–3378 CAS.
- J. Mahieux, S. Gonella, M. Sanselme and G. Coquerel, CrystEngComm, 2012, 14, 103–111 RSC.
- F. Gagiu, C. Nistor, F. Danciu, A. Cacoveanu, G. Luputiu, C. Ban and Gh. Hintz, Ann. Pharm. Fr., 1973, 31, 363–367 CAS.
- G. A. Morales and F. R. Fronczek, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 52, 1266–1268 CrossRef.
- I. Bernal, F. Somoza and V. Bahn, J. Coord. Chem., 1997, 42, 1–10 CrossRef CAS.
- J. Cai, J. Myrczek, H. Chun and I. Bernal, J. Chem. Soc., Dalton Trans., 1998, 4155–4160 RSC.
- L. Fábián and C. P. Brock, Acta Crystallogr., Sect. B: Struct. Sci., 2010, 66, 94–103 Search PubMed.
- I. Bernal and S. Watkins, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 216–221 CAS.
- R. Laubenstein, M.-D. erb, U. Englert, G. Raabe, T. Braun and B. Braun, Chem. Commun., 2016, 52, 1214–1217 RSC.
- P. S. Carvalho Jr, J. Ellena, D. S. Yufit and J. A. K. Howard, Cryst. Growth Des. DOI:10.1021/acs.cgd.6b00442.
- M. C. Etter, Acc. Chem. Res., 1990, 23, 120–126 CrossRef CAS.
- J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555–1573 CrossRef CAS.
- K. Suresh, N. R. Goud and A. Nangia, Chem. – Asian J., 2013, 8, 3032–3041 CrossRef CAS PubMed.
- S. Cherukuvada, G. Bolla, K. Sikligar and A. Nangia, Cryst. Growth Des., 2013, 13, 1551–1557 CAS.
- M. G. Smith, R. P. Forbes and A. Lemmerer, Cryst. Growth Des., 2015, 15, 3813–3821 CAS.
- Y. Mnyukh, Am. J. Condens. Matter Phys., 2013, 3, 89–103 Search PubMed.
- H. Ito, M. Muromoto, S. Kurenuma, S. Ishizaka, N. Kitamura, H. Sato and T. Seki, Nat. Commun., 2013, 4, 2009 Search PubMed.
- S. Kobatake, S. Takami, H. Muto, T. Ishikawa and M. Irie, Nat. Commun., 2007, 446, 778–781 CrossRef CAS PubMed.
- T. Mutai, H. Satou and K. Araki, Nat. Mater., 2005, 4, 685–687 CrossRef CAS PubMed.
- M. Lusi and J. Bernstein, Chem. Commun., 2013, 49, 9293–9295 RSC.
- W. Ostwald, Z. Phys. Chem., 1879, 22, 289–330 Search PubMed.
- T. Threlfall, Org. Process Res. Dev., 2003, 7, 1017–1027 CrossRef CAS.
- S.-Y. Chung, Y.-M. Kim, J.-G. Kim and Y.-J. Kim, Nat. Phys., 2009, 5, 68–73 CrossRef CAS.
- R. K. Hylton, G. J. Tizzard, T. L. Threlfall, A. L. Ellis, S. J. Coles, C. C. Seaton, E. Schulze, H. Lorenz, A. Seidel-Morgenstern, M. Stein and S. L. Price, J. Am. Chem. Soc., 2015, 137, 11095–11104 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and analytical data. CCDC 1480079–1480081. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc04148e |
| This journal is © The Royal Society of Chemistry 2016 |