
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic enantio- and diastereoselective Mannich reaction of α-substituted isocyanoacetates and ketimines†
Raquel
de la Campa
,
Adam D.
Gammack Yamagata
,
Irene
Ortín
,
Allegra
Franchino
,
Amber L.
Thompson
,
Barbara
Odell
and
Darren J.
Dixon
*
Chemistry Research Laboratory, Department of Chemistry, University of Oxford, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: darren.dixon@chem.ox.ac.uk
First published on 1st August 2016
Abstract
The highly diastereo- and enantioselective Mannich addition/cyclisation reaction of α-substituted isocyanoacetate ester pronucleophiles and (hetero)aryl and alkyl methyl ketone-derived ketimines using a silver acetate and a cinchona-derived amino phosphine binary catalyst system is reported.
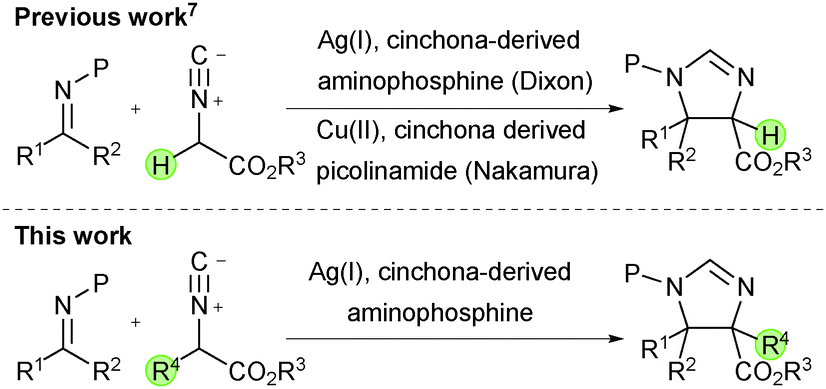
The Mannich reaction is a powerful tool for the creation of carbon–carbon bonds1 and since its discovery in the early 20th century great progress in the development of catalytic enantioselective variants has been made.2 To date, the majority of studies have employed aldimines for the enantioselective preparation of amine derivatives possessing tertiary stereogenic centres.3,4 However recent efforts have focussed on the development of new catalytic Mannich reactions with ketimines for the stereoselective production of amine derivatives possessing tetrasubstituted stereogenic centres;5 the construction of which remains an important challenge in the field.6 To this end, our group recently developed a catalytic enantioselective Mannich reaction of isocyanoacetate esters and ketimines to generate trisubstituted chiral imidazolines possessing vicinal quaternary and tertiary stereogenic carbon atoms.7 Under the control of a binary catalyst system comprising silver oxide and a cinchona alkaloid-derived amino phosphine, impressive reactivity and high enantio- and diastereocontrol were achieved.8 As well as being important building blocks for the synthesis of α,β-diamino acids,9 substituted imidazolines, such as those produced by this chemistry, can be identified as the core of biologically active compounds.10
We recognised that the catalytic enantioselective Mannich reaction of α-substituted isocyanoacetate pronucleophiles with ketimines could provide a synthetically powerful approach for the enantioselective synthesis of imidazolines possessing two vicinal tetrasubstituted stereocentres.11 To this end, and in a further development of our ongoing research program, we wish to report our efforts leading to a catalytic enantioselective ketimine Mannich reaction of α-substituted isocyanoacetate esters affording chiral imidazolines with two contiguous fully substituted carbon stereocentres, promoted by a catalytic system made from a cinchona-derived amino phosphine ligand and a silver(I) salt (Scheme 1).
 | ||
| Scheme 1 Enantioselective construction of chiral imidazolines with two contiguous tetrasubstituted stereocentres. | ||
Initially we selected acetophenone-derived N-diphenylphosphinoyl ketimine 3a as our model electrophile and investigated a range of 2-isocyanopropanoate ester pronucleophiles 2 using our previously optimised7a catalyst system comprising quinine-derived amino phosphine 1a (20 mol%) and Ag2O (5 mol%) in ethyl acetate at −20 °C (Table 1). Methyl 2-isocyanopropanoate 2a was first investigated as the pronucleophile. Very pleasingly after 60 hours, N-DPP protected trans-imidazoline 4a was afforded as a single diastereomer. To allow full characterisation and determination of enantiomeric excess (ee) via HPLC analysis, DPP group removal using 1 M HCl was found to be necessary and imidazoline 5a was afforded in 59% yield over the two steps, in 89% ee (entry 1). With reasonable reactivity and stereoselectivity identified, the performance of more bulky 2-isocyanopropanoate esters – specifically iso-propyl (2b), tert-butyl (2c), benzyl (2d) and benzhydryl (2e) 2-isocyanopropanoates – was studied (entries 2–5). With tert-butyl ester 2c the high enantioselectivity (89% ee) was maintained and the reaction yield was slightly improved to 63% (entry 3), therefore subsequent optimisation in terms of catalyst loading, reactivity and enantiocontrol was done using this pronucleophile. The loading with respect to amino phosphine 1 was lowered to 10 mol% and that of the Ag(I) ions to 5 mol% and the effect of the silver salt was investigated. Interestingly, when the reaction was run using Ag2O in the presence of 1a the yield increased to 85% and enantioselectivity was maintained at 89% ee (entry 6). With silver carbonate a marginal increase in enantioselectivity (to 90% ee) was seen but the yield was lower (75%, entry 7). With both Ag2O and Ag2CO3 the formation of a translucent precipitate was observed. Efforts to form a more soluble complex were made by changing the silver source to AgOAc,12 which pleasingly gave imidazoline 5c in high ee (89%) with an increased yield (90%) within a shorter reaction time (48 hours, entry 8). Quinidine-derived ligand 1b and cinchonidine-derived amino phosphine 1c with silver acetate afforded 5c in slightly lower enantioselectivity (entries 9 and 10, 80% and 86% ee respectively). Increasing the reaction temperature to 0 °C had a negligible effect on both ee and yield, whereas decreasing it to −30 °C completely suppressed reactivity (entries 11 and 12). Control experiments, first omitting 1a and secondly AgOAc, confirmed that the silver salt was critical for reactivity, whilst the amino phosphine was essential for stereocontrol and important for a high yield (entries 13 and 14).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | 1 (mol%) | [Ag] | R1 | 2 | 5 | t (h) | Yieldb | eec |
a Performed under N2 with 1 equiv. of 2, 1.1 equiv. of 3a, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ag(I) in 2:1 ratio, 4 Å MS in AcOEt (0.09 M) at −20 °C.
b Yield over two steps for isolated product 5 (single diastereomer) after flash column chromatography (FCC).
c Enantiomeric excess (ee) of compound 5 determined by HPLC analysis on a chiral stationary phase.
d Enantiomeric (4R,5S)-5c obtained.
e At 0 °C.
f At −30 °C.
g With 10 mol% AgOAc and 10 mol% 1a. :Ag(I) in 2:1 ratio, 4 Å MS in AcOEt (0.09 M) at −20 °C.
b Yield over two steps for isolated product 5 (single diastereomer) after flash column chromatography (FCC).
c Enantiomeric excess (ee) of compound 5 determined by HPLC analysis on a chiral stationary phase.
d Enantiomeric (4R,5S)-5c obtained.
e At 0 °C.
f At −30 °C.
g With 10 mol% AgOAc and 10 mol% 1a.
|
||||||||
| 1 | 1a (20) | Ag2O | Me | 2a | 5a | 60 | 59 | 89 |
| 2 | 1a (20) | Ag2O | iPr | 2b | 5b | 60 | 77 | 79 |
| 3 | 1a (20) | Ag2O | t Bu | 2c | 5c | 60 | 63 | 89 |
| 4 | 1a (20) | Ag2O | Bn | 2d | 5d | 60 | 77 | 83 |
| 5 | 1a (20) | Ag2O | CHPh2 | 2e | 5e | 60 | 62 | 84 |
| 6 | 1a (10) | Ag2O | t Bu | 2c | 5c | 60 | 85 | 89 |
| 7 | 1a (10) | Ag2CO3 | t Bu | 2c | 5c | 70 | 75 | 90 |
| 8 | 1a (10) | AgOAc | t Bu | 2c | 5c | 48 | 90 | 89 |
| 9 | 1b (10) | AgOAc | t Bu | 2c | 5c | 48 | 81 | 80d |
| 10 | 1c (10) | AgOAc | t Bu | 2c | 5c | 48 | 89 | 86 |
| 11e | 1a (10) | AgOAc | t Bu | 2c | 5c | 48 | 82 | 88 |
| 12f | 1a (10) | AgOAc | t Bu | 2c | 5c | 120 | 0 | n/a |
| 13 | — | AgOAc | t Bu | 2c | 5c | 72 | 64 | 0 |
| 14 | 1a (10) | — | t Bu | 2c | 5c | 120 | 0 | n/a |
| 15 | 1a (2.5) | AgOAc | t Bu | 2c | 5c | 72 | 89 | 90 |
| 16g | 1a (10) | AgOAc | t Bu | 2c | 5c | 48 | 75 | 90 |
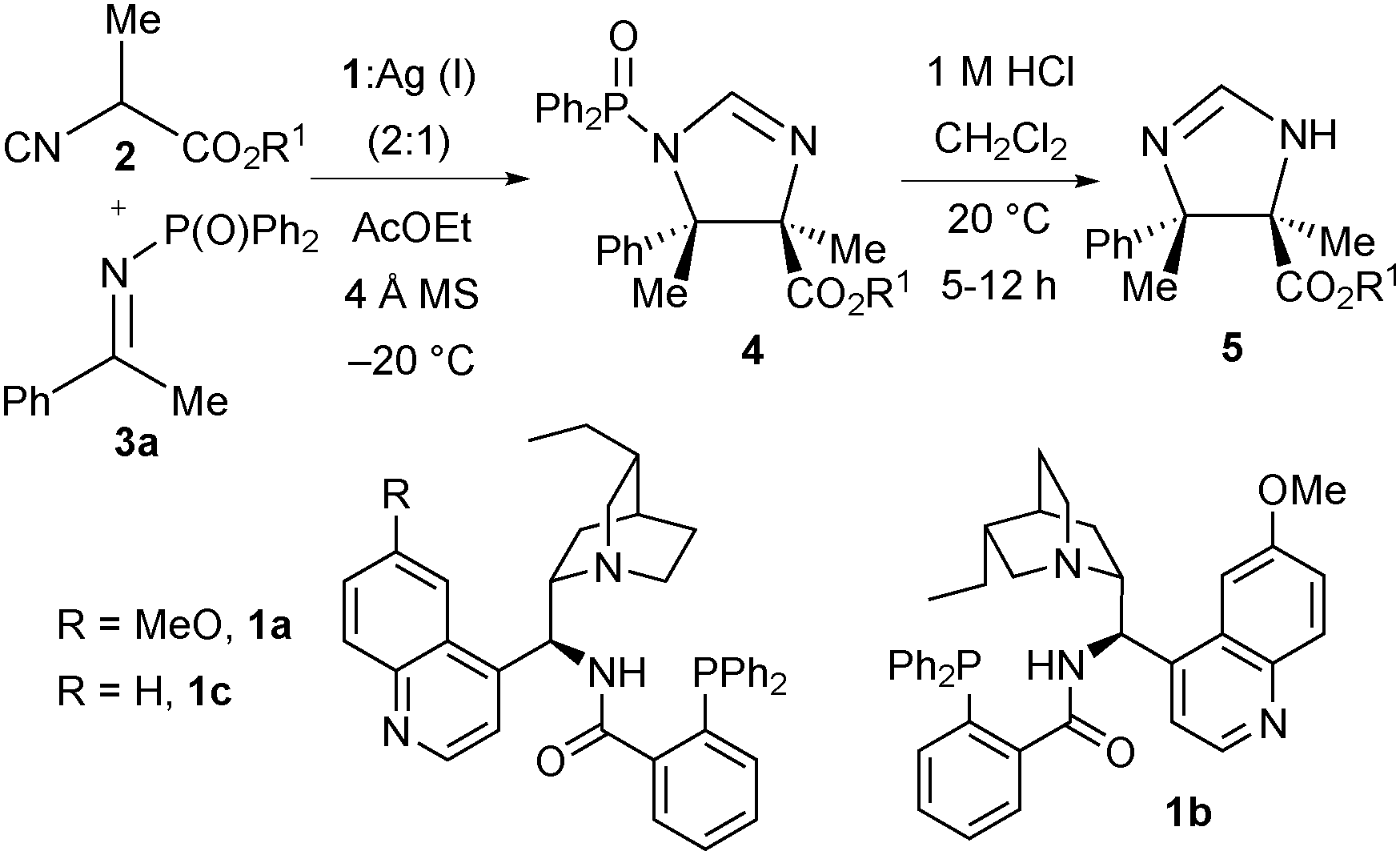
It was possible to further reduce the ligand:AgOAc loading to 2.5:1.25 mol% with no effect on the ee or yield albeit with slightly increased reaction time (entry 15). A 1:1 ratio of ligand:AgOAc resulted in slightly lower yield (entry 16). For convenience in weighing out the small amounts of silver acetate, the scope of the reaction was assessed at 10:5 mol% ligand:AgOAc ratio at −20 °C.
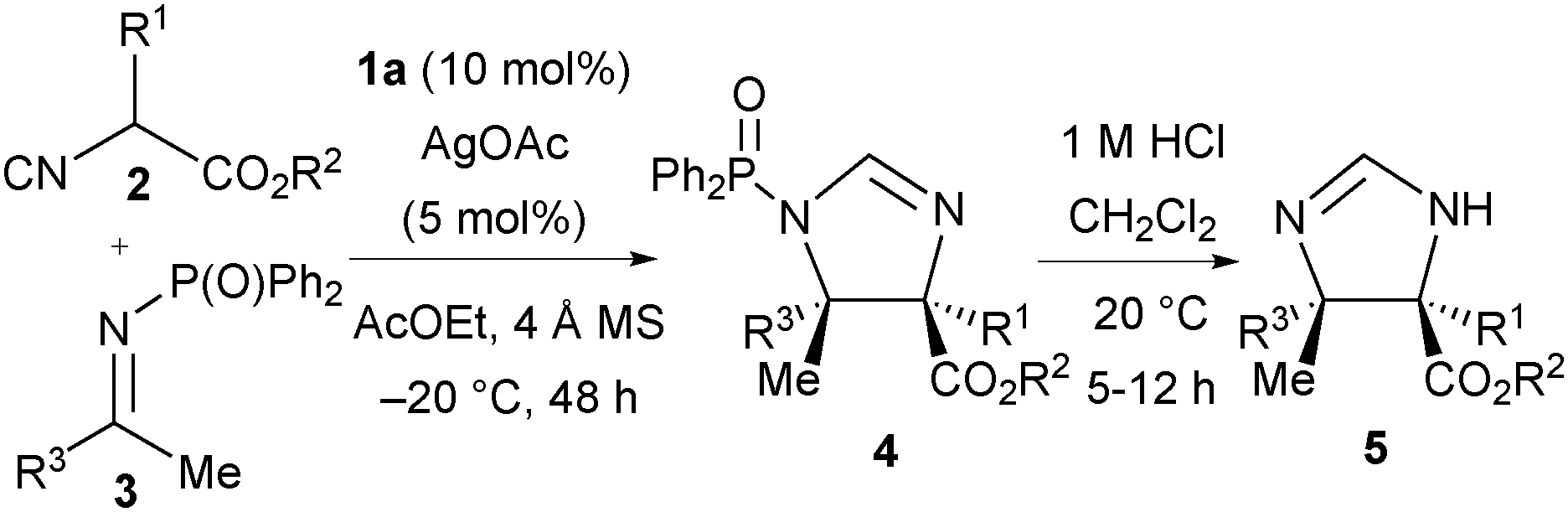
A range of ketimines derived from aromatic and heteroaromatic methyl ketones13 (3a–3j) were subjected to the reaction conditions (Table 2). Electron-withdrawing and electron-donating groups in the 4-position of the aromatic ring were well tolerated, giving 5f–5j in good yield (71–89%) and enantiomeric excess (88–90% ee, entries 2–6). Meta- and ortho-substituted aromatic ketimines afforded the imidazolines 5k and 5l with lower yield (65% and 63% respectively) and good ee (87% and 95% respectively, entries 7 and 8). Heteroaromatic N-DPP ketimines derived from methyl 3-pyridyl ketone and methyl 2-furyl ketone gave products 5m and 5n in high yield and ee (entries 9 and 10). Aliphatic ketimine 3k derived from 4-phenyl-2-butanone could be converted to imidazoline 5o with moderate yield (48%) and good enantioselectivity (87% ee, entry 11). Next, tert-butyl 2-isocyanobutanoate 2f was assessed on a range of ketimines to afford exclusively the trans imidazolines 5p–5s with an ethyl substituent in the α-position in similarly high ee and yield (entries 12–15). The tert-butyl 2-isocyanohexanoate 2g also performed well in the reaction to give products 5t–5w in excellent yield and ee (entries 16–19).14 NOe NMR experiments established the relative trans stereochemical configuration of products 5f, 5o, 5q and 5v. Absolute configuration was determined by comparison of the specific rotation of imidazoline 5y with that reported in the literature.5c
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | R3 | 3 | R1 | R2 | 2 | 5 | Yieldb | ee |
|
a Performed under N2 with 1 equiv. of 2, 1.1 equiv. of 3, 0.1 equiv. of 1a, 0.05 equiv. of AgOAc, 4 Å MS in AcOEt (0.09 M) at −20 °C for 48 h. Imidazolines 4 were obtained with dr > 98:2 (determined by 1H-NMR analysis of the crude reaction mixture) unless otherwise stated.
b Yield over two steps for isolated product 5 (single diastereomer) after FCC.
c
N-DPP imidazoline 4o was obtained with a 5:1 dr in favour of the trans isomer, as determined by 1H-NMR analysis of the crude reaction mixture.
|
||||||||
| 1 | Ph | 3a | Me | t Bu | 2c | 5c | 90 | 89 |
| 2 | 4-MeC6H4 | 3b | Me | t Bu | 2c | 5f | 71 | 89 |
| 3 | 4-PhC6H4 | 3c | Me | t Bu | 2c | 5g | 87 | 88 |
| 4 | 4-FC6H4 | 3d | Me | t Bu | 2c | 5h | 89 | 90 |
| 5 | 4-ClC6H4 | 3e | Me | t Bu | 2c | 5i | 80 | 89 |
| 6 | 4-MeOC6H4 | 3f | Me | t Bu | 2c | 5j | 80 | 90 |
| 7 | 3-ClC6H4 | 3g | Me | t Bu | 2c | 5k | 65 | 87 |
| 8 | 2-FC6H4 | 3h | Me | t Bu | 2c | 5l | 63 | 95 |
| 9 | 3-Pyridyl | 3i | Me | t Bu | 2c | 5m | 80 | 87 |
| 10 | 2-Furyl | 3j | Me | t Bu | 2c | 5n | 83 | 91 |
| 11 | PhCH2CH2 | 3k | Me | t Bu | 2c | 5o | 48c | 87 |
| 12 | Ph | 3a | Et | t Bu | 2f | 5p | 62 | 90 |
| 13 | 4-ClC6H4 | 3e | Et | t Bu | 2f | 5q | 81 | 89 |
| 14 | 4-MeOC6H4 | 3f | Et | t Bu | 2f | 5r | 79 | 92 |
| 15 | 2-Furyl | 3j | Et | t Bu | 2f | 5s | 84 | 93 |
| 16 | Ph | 3a | n Bu | t Bu | 2g | 5t | 85 | 91 |
| 17 | 4-FC6H4 | 3d | n Bu | t Bu | 2g | 5u | 83 | 90 |
| 18 | 4-ClC6H4 | 3e | n Bu | t Bu | 2g | 5v | 80 | 88 |
| 19 | 2-Furyl | 3j | n Bu | t Bu | 2g | 5w | 83 | 93 |
| 20 | 4-MeC6H4 | 3b | Me | Me | 2a | 5y | 28 | 87 |
As a demonstration of the synthetic utility of this newly established method, product 5c was hydrolysed to afford α,β-diamino acid 6c (Scheme 2). Such compounds are difficult to access in enantioenriched form by other methods.9
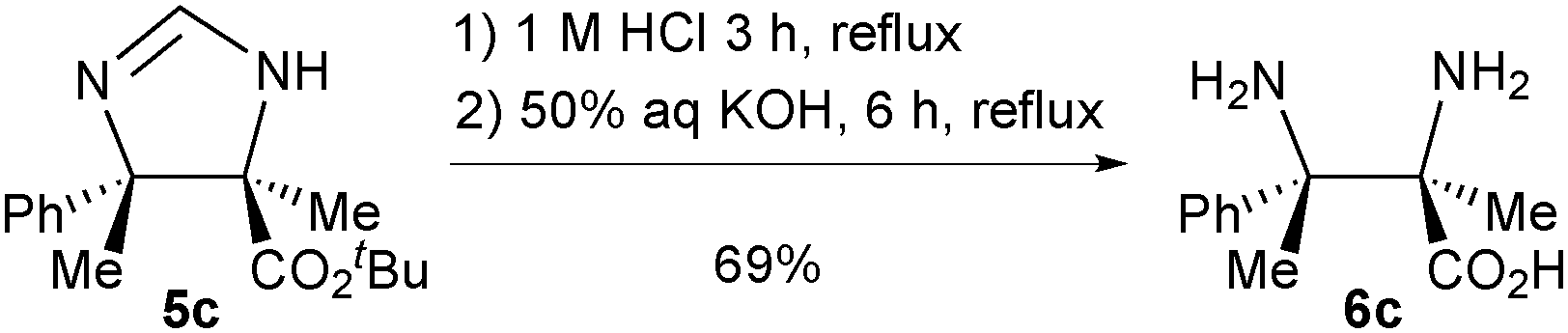 | ||
| Scheme 2 Application to the synthesis of fully substituted α,β-diamino acids. | ||
To gain insight into the importance of the various functional groups possessed by amino phosphines 1, a series of control experiments using modified ligand structures (1d–1g) were performed (Table 3). Diastereomeric ligand 1d gave imidazoline 5c with a large decrease in enantioselectivity (entry 2). Ligand 1e, lacking the phosphine moiety, was significantly worse: product 5c was obtained in low yield with poor enantioselectivity (entry 3). Ligand 1f with an ester linker gave imidazoline 5c in slightly decreased yield and enantioselectivity (entry 4). Finally ligand 1g with a N-methyl group on the amide promoted the reaction with a similar yield to 1a but much lower levels of enantioselectivity (entry 5).
In order to better understand the results from the catalyst modification studies and to determine how ligand 1a binds to AgOAc, a crystal of complex 1a·AgOAc was grown (Fig. 1).15 Single crystal X-ray diffraction determined that in the solid state 1a acts as a P,N-bidentate ligand, binding silver through both the phosphine and the quinuclidine N atom to create a 9-membered macrocycle. The other groups coordinating to silver are the acetate and a quinoline unit belonging to a symmetry related molecule of 1a leading to the formation of polymeric chains in the solid state. The four-coordinate silver adopts a distorted tetrahedral geometry. An intramolecular hydrogen bond between the acetate group on silver and the transannular amide N–H at 1.896(3) Å is present with a N⋯O distance of 2.752(4) Å. The π system of the amide group is oriented towards the silver with a distance of 2.9571(18) Å between the amide nitrogen and the silver centre suggesting a possible interaction. Such an interaction would transfer charge resulting in acidification of the amide NH thus enhancing its H-bond donor ability.
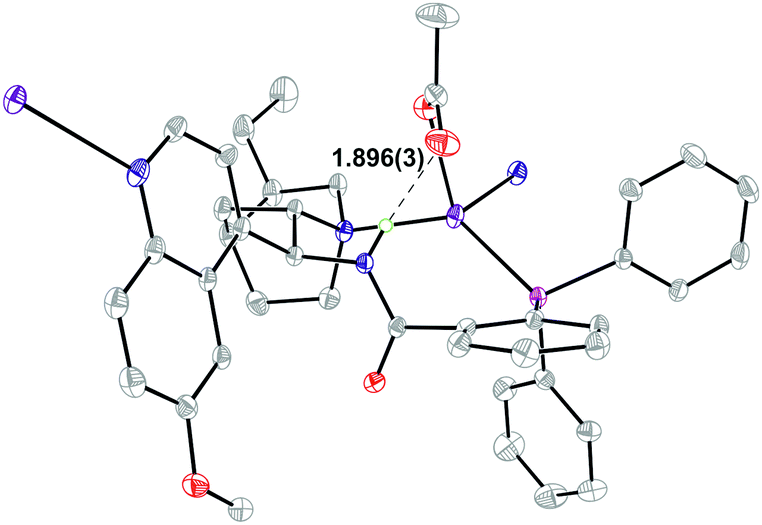 | ||
| Fig. 1 Structure of 1a·AgOAc from single crystal X-ray diffraction studies showing the intramolecular H-bond (H⋯O = 1.896(3) Å; N⋯O = 2.752(4) Å). Displacement ellipsoids drawn at 30% probability; solvent and selected hydrogen atoms omitted for clarity. | ||
31P and 1H NMR studies of a 1:1 ratio of amino phosphine 1a and AgOAc at −20 °C were also performed (see ESI† for full NMR studies). The 31P NMR experiments showed a quantitative binding of silver(I) in solution and 1H NMR analysis of the same mixture revealed a large downfield shift (approximately 3.4 ppm relative to that of the free ligand) of the amide N–H consistent with presence of the same H-bonding interaction. Additionally, the protons adjacent to the quinuclidine nitrogen were also shifted downfield, consistent with coordination of the quinuclidine nitrogen to silver. In the solution phase there is no strong evidence that the quinoline N atom binds the Ag centre; the change of chemical shift for the proton adjacent to the quinoline nitrogen is only 0.07 ppm. When methyl isocyanoacetate is added to the solution of 1a and AgOAc, the CH2 group appears as a characteristic ABq system, providing evidence for complexation of the isonitrile to silver.
Building on the results of experiments using modified ligand structures (Table 3) and the structural data obtained from both the X-ray and NMR studies we propose a transition structure that rationalises the enantio- and diastereoselectivity observed in this Mannich reaction (Fig. 2). The N atom of the (E)-configurated ketimine is postulated to make a key hydrogen bond to the amide N–H, which both orientates the electrophile and activates it towards nucleophilic addition. The aromatic ring of the H-bonded ketimine is oriented away from the quinuclidine and quinoline units on steric grounds and C–H/π-interactions16 between it and the aromatic ring of the carboxamide could provide additional binding/organisation, possibly accounting for the high enantiofacial selectivity observed using this catalyst.7a,8 The ester group of the isocyanoacetate is oriented away from the aromatic group on the ketimine on steric grounds. Subsequent to the enantio- and diastereodetermining C–C bond forming step, ring closure and protonolysis would yield the N-DPP protected imidazoline with the observed stereochemical configuration. Satisfyingly, this transition state model, with its multipoint binding interactions, would also rationalise the stereochemical outcomes of the previously reported reactions with aldehydes8a and ketones.8b
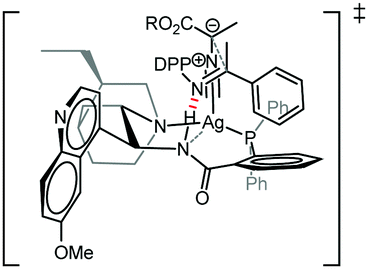 | ||
| Fig. 2 Proposed transition structure. | ||
In conclusion, we have developed a diastereo- and enantioselective Mannich reaction/cyclisation of α-substituted isocyanoacetates and ketimines catalysed by silver acetate and a cinchona-derived amino phosphine. The reaction scope is broad and includes substituted aryl, heteroaryl and alkyl methyl ketimines as well as isocyanoacetates with linear alkyl chains in the α-position. Hydrolysis of the product imidazolines provides access to fully substituted α,β-diamino acids. Information arising from X-ray and NMR studies, as well as control studies on the ligand, have allowed a detailed transition state model to be postulated.
The authors thank the EPSRC for a leadership fellowship to D. J. D., Merck, Sharp & Dohme (Hoddesdon, UK) for a CASE award to A. D. G. Y., and the EC for IEF to R. C. (PIEF-GA-2012-329689), IEF to I. O. (PIEF-GA-2010-275788) and PhD fellowship to A. F. (FP7/2007-2013, REA grant agreement 316955).
Notes and references
- T. Akiyama, in Comprehensive Organic Synthesis, ed. P. Knochel and G. A. Molander, Elsevier, Oxford, 2nd edn, 2014, vol. 2, p. 629 Search PubMed.
- (a) S. Kobayashi and H. Ishitani, Chem. Rev., 1999, 99, 1069 CrossRef CAS PubMed; (b) A. Cordova, Acc. Chem. Res., 2004, 37, 102 CrossRef CAS PubMed; (c) N. Z. Burns and E. N. Jacobsen, in Science of Synthesis, Stereoselective Synthesis, ed. J. G. De Vries, G. A. Molander and P. A. Evans, Thieme, Stuttgart, 2011, vol. 2, p. 785 Search PubMed.
- Enantioselective Mannich reactions of aldimines: (a) K. Juhl, N. Gathergood and K. A. Jørgensen, Angew. Chem., Int. Ed., 2001, 40, 2995 CrossRef CAS; (b) L. Bernardi, A. S. Gothelf, R. G. Hazell and K. A. Jørgensen, J. Org. Chem., 2003, 68, 2583 CrossRef CAS PubMed; (c) T. Ooi, M. Kameda, J.-I. Fujii and K. Maruoka, Org. Lett., 2004, 6, 2397 CrossRef CAS PubMed; (d) A. Okada, T. Shibuguchi, T. Ohshima, H. Masu, K. Yamaguchi and M. Shibasaki, Angew. Chem., Int. Ed., 2005, 44, 4564 CrossRef CAS PubMed; (e) N. S. Chowdari, M. Ahmad, K. Albertshofer, F. Tanaka and C. F. Barbas III, Org. Lett., 2006, 8, 2839 CrossRef CAS PubMed; (f) S. Kobayashi, R. Yazaki, K. Seki and Y. Yamashita, Angew. Chem., Int. Ed., 2008, 47, 5613 CrossRef CAS PubMed; (g) J. Hernández-Toribio, R. G. Arrayás and J. C. Carretero, J. Am. Chem. Soc., 2008, 130, 16150 CrossRef PubMed; (h) D. Shang, Y. Liu, X. Zhou, X. Liu and X. Feng, Chem. – Eur. J., 2009, 15, 3678 CrossRef CAS PubMed; (i) J. Hernández-Toribio, R. G. Arrayás and J. C. Carretero, Chem. – Eur. J., 2010, 16, 1153 CrossRef PubMed; (j) G. Liang, M.-C. Tong, H. Tao and C.-J. Wang, Adv. Synth. Catal., 2010, 352, 1851 CrossRef CAS; (k) E. Hernando, R. G. Arrayás and J. C. Carretero, Chem. Commun., 2012, 48, 9622 RSC.
- Enantioselective Mannich reactions of isocyanoacetate esters and aldimines: (a) X.-T. Zhou, Y.-R. Lin, L.-X. Dai, J. Sun, L.-J. Xia and M.-H. Tang, J. Org. Chem., 1999, 64, 1331 CrossRef CAS; (b) X.-T. Zhou, Y.-R. Lin and L.-X. Dai, Tetrahedron: Asymmetry, 1999, 10, 855 CrossRef CAS; (c) J. Aydin, A. Rydén and K. J. Szabó, Tetrahedron: Asymmetry, 2008, 19, 1867 CrossRef CAS; (d) Z.-W. Zhang, G. Lu, M.-M. Chen, N. Lin, Y.-B. Li, T. Hayashi and A. S. C. Chan, Tetrahedron: Asymmetry, 2010, 21, 1715 CrossRef CAS; (e) S. Nakamura, Y. Maeno, M. Ohara, A. Yamamura, Y. Funanashi and N. Shibata, Org. Lett., 2012, 14, 2960 CrossRef CAS PubMed; (f) P.-L. Shao, J.-Y. Liao, Y. A. Ho and Y. Zhao, Angew. Chem., Int. Ed., 2014, 53, 5435 CrossRef CAS PubMed.
- Enantioselective Mannich reactions of ketimines: (a) Y. Du, L.-W. Xu, Y. Shimizu, K. Oisaki, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2008, 130, 16146 CrossRef CAS PubMed; (b) L. C. Wieland, E. M. Vieira, M. L. Snapper and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 570 CrossRef CAS PubMed; (c) G. Lu, T. Yoshino, H. Morimoto, S. Matsunaga and M. Shibasaki, Angew. Chem., Int. Ed., 2011, 50, 4382 CrossRef CAS PubMed; (d) M. Hayashi, M. Sano, Y. Funahashi and S. Nakamura, Angew. Chem., Int. Ed., 2013, 52, 5557 CrossRef CAS PubMed; (e) M. G. Núñez, A. J. M. Farley and D. J. Dixon, J. Am. Chem. Soc., 2013, 135, 16348 CrossRef PubMed; (f) Y.-Q. Wang, Y. Zhang, K. Pan, J. You and J. Zhao, Adv. Synth. Catal., 2013, 355, 3381 CrossRef CAS; (g) M.-X. Zhao, H.-L. Bi, R.-H. Jiang, X.-W. Xu and M. Shi, Org. Lett., 2014, 16, 4566 CrossRef CAS PubMed; (h) S. Zhang, L. Li, Y. Hu, Z. Zha, Z. Wang and T.-P. Loh, Org. Lett., 2015, 17, 1050 CrossRef CAS PubMed; (i) S. Lin, Y. Kawato, N. Kumagai and M. Shibasaki, Angew. Chem., Int. Ed., 2015, 54, 5183 CrossRef CAS PubMed; (j) S. Nakamura, R. Yamaji and M. Hayashi, Chem. – Eur. J., 2015, 21, 9615 CrossRef CAS PubMed; (k) X. Liu, J. Zhang, L. Zhao, S. Ma, D. Yang, W. Yan and R. Wang, J. Org. Chem., 2015, 80, 12651 CrossRef CAS PubMed; (l) S. Lin, N. Kumagai and M. Shibasaki, Chem. – Eur. J., 2016, 22, 3296 CrossRef CAS PubMed; (m) T. Takeda, A. Kondoh and M. Terada, Angew. Chem., Int. Ed., 2016, 55, 4734 CrossRef CAS PubMed.
- J. Christoffers and A. Mann, Angew. Chem., Int. Ed., 2001, 40, 4591 CrossRef CAS.
- (a) I. Ortín and D. J. Dixon, Angew. Chem., Int. Ed., 2014, 53, 3462 CrossRef PubMed; (b) M. Hayashi, M. Iwanaga, N. Shiomi, D. Nakane, H. Masuda and S. Nakamura, Angew. Chem., Int. Ed., 2014, 53, 8411 CrossRef CAS PubMed.
- (a) F. Sladojevich, A. Trabocchi, A. Guarna and D. J. Dixon, J. Am. Chem. Soc., 2011, 133, 1710 CrossRef CAS PubMed; (b) R. de la Campa, I. Ortín and D. J. Dixon, Angew. Chem., Int. Ed., 2015, 54, 4895 CrossRef CAS PubMed.
- A. Viso, R. Fernández de la Pradilla, M. Tortosa, A. García and A. Flores, Chem. Rev., 2011, 111, PR1 CrossRef PubMed.
- H. Liu and D.-M. Du, Adv. Synth. Catal., 2009, 351, 489 CrossRef CAS.
- During the preparation of the present manuscript, the following article was published online: S. Nakamura, R. Yamaji and M. Iwanaga, Chem. Commun., 2016, 52, 7462 RSC.
- Q.-A. Chen, D.-S. Wang and Y.-G. Zhou, Chem. Commun., 2010, 46, 4043 RSC.
- Reaction between 2c and 4′-bromopropiophenone-derived ketimine, followed by removal of the DPP group, gave the corresponding imidazoline in 62% overall yield and 70% ee.
- Reaction between 3a and valine-derived tert-butyl isocyanoacetate (isopropyl group at the α-position), followed by DPP deprotection, gave the corresponding imidazoline in 29% overall yield (8 days reaction time) and 98% ee.
- CCDC 1493438.
- S. Tsuzuki and A. Fujii, Phys. Chem. Chem. Phys., 2008, 10, 2584 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Procedures, characterisation data, copies of NMR spectra, HPLC traces, X-ray data and CIF file. CCDC 1493438. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc04132a |
| This journal is © The Royal Society of Chemistry 2016 |