
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supersizing pyrrole-modified porphyrins by reversal of the ‘breaking and mending’ strategy†
M.
Luciano
a,
W.
Tardie
a,
M.
Zeller
b and
C.
Brückner
*a
aDepartment of Chemistry, University of Connecticut, 55 N. Eagleville Rd., Storrs, CT 06269-3060, USA. E-mail: c.bruckner@uconn.edu
bDepartment of Chemistry, Purdue University, 560 Oval Drive, West Lafayette, IN 47907-2084, USA
First published on 19th July 2016
Abstract
While the ‘breaking and mending of porphyrin strategy’ proved versatile in the generation of a range of pyrrole-modified porphyrins containing 4-, 5-, and 6-membered heterocycles, it failed to access systems incorporating larger rings. A reversal of the strategy – first mending, then breaking – now allowed the formation of a pyrrole-modified porphyrin containing an 8-membered 1,3,6-triazocine-2,4,8-trione heterocycle.
The synthesis of porphyrin analogues has contributed to the understanding of the concept of aromaticity,1 provided macrocycles with valuable molecular recognition or catalytic properties,2 and has provided a large number of dyes with optical properties that are inaccessible with regular porphyrins or hydroporphyrins.3 The majority of porphyrin analogues have been prepared by total synthesis.4 In an approach we dubbed ‘the breaking and mending of porphyrins’, we,5 and others,4e prepared a wide variety of porphyrin analogues containing one or two non-pyrrolic heterocycles by step-wise conversion of a porphyrin. To illustrate the approach, meso-tetraphenylporphyrin 1, for example, was dihydroxylated; the diol functionality was then used as a synthetic handle for further functionalizations (Scheme 1).5 Diol cleavage in the ‘breaking’ step resulted in the formation of a secochlorin 3 that could be cyclized in the ‘mending’ step to, e.g., provide a morpholinochlorin incorporating a 6-membered morpholine moiety.6 Attempts to generate 7-membered rings (1,4,5-triazaazepines or 1,3-diaza-5-oxaazepines) by cyclization of 3 with hydrazine or hydroxylamine, respectively, failed.7 In all likelihood, the analogues formed but then rapidly extruded a small molecule (N2 or CO2, respectively), regenerating the porphyrin or a porphyrin-like arrangement of four five-membered rings. This highlights the large stability of the natural porphyrinic architecture, but also identifies a limitation of our ‘breaking and mending’ approach toward pyrrole-modified porphyrins containing medium-sized rings.
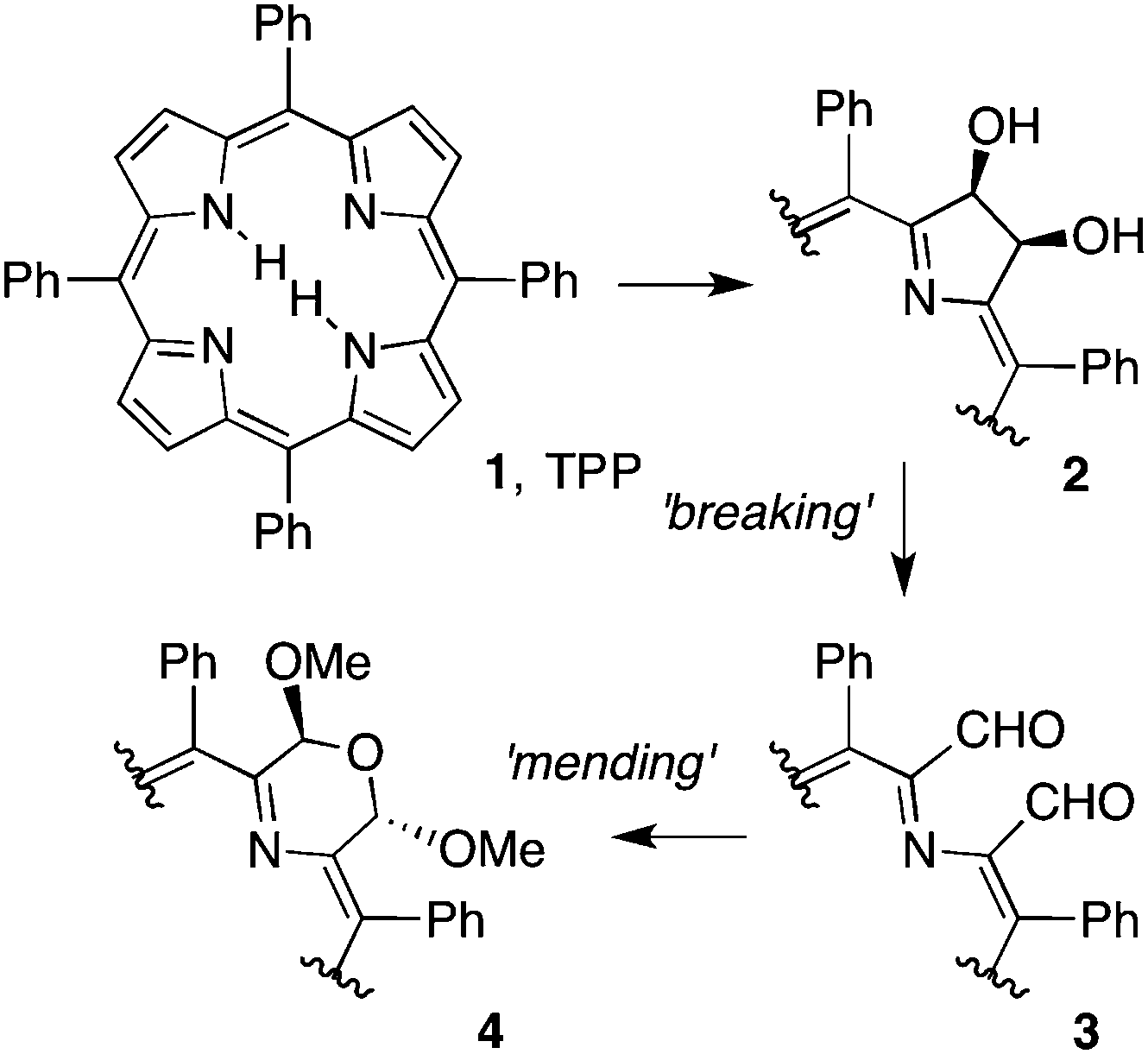 | ||
| Scheme 1 Example for the ‘breaking and mending of porphyrins’ approach toward porphyrinoids containing non-pyrrolic building blocks. | ||
Albeit rare, porphyrinoids prepared by total synthesis containing 7-membered rings are stable.4e The best investigated systems are the tropiporphyrins, a family of carbaporphyrins.4d Azepiphthalocyanines were also reported.8 Medium-sized rings can, in principle, be accessed by the cleavage of the linkage between two annulated smaller rings.9 In the realm of porphyrinoid chemistry, a single such example was provided in 2003 by the groups of Barrett and Hoffman (Scheme 2).10 Diazapine-annulated porphyrazine 5 was prepared by total synthesis. Subsequently, the β,β′-bond linking the two heterocycles was cleaved, forming the pyrrole-expanded porphyrazine 6 containing a 10-membered heterocycle.
 | ||
| Scheme 2 Synthesis of pyrrole-expanded porphyrazine 6 described by the groups of Barrett and Hoffman. Reaction conditions: (i) KMnO4, CH2Cl2 (44% yield).10 | ||
With this precedent in mind, we set out to test whether a reversal of our traditional methodology, i.e., a ‘mending and breaking’ approach can be developed into a possibly general approach toward the synthesis of pyrrole-modified porphyrins with medium-sized heterocyclic rings that can also take advantage of some of the reaction types well established by us. This contribution will demonstrate that this is indeed the case, but it will also point at some limitations to this approach.
meso-Tetraarylporphyrin diones, such as 7, are accessible from TPP (1) along a number of complimentary routes (Scheme 3).11 The regular ketone reactivity of the diones was previously demonstrated,11b including in reactions generating pyrrole-modified porphyrins.12 It is well-known that reaction of dione 7 with diamines generates diimines,13 but we found that the reaction of 7 with urea or N,N′-dimethylurea generated dihydroxychlorins 8 and 9, respectively, containing imidazolidinone moieties annulated at the β,β′-positions.‡ Diagnostic for the formation of the dihydroxychlorin structure are the preservation of the two-fold symmetry of the adducts as seen in their NMR spectra, the pyrroline carbon signals in their 13C NMR spectra (found at δ = 158.5 ppm for 8), their regular chlorin-like spectra compared to the much broadened spectra for dione 714 (Fig. 1A), and their expected composition (as per HR-MS). The presence of the lactam moieties in the annulated ring are indicated in the 13C NMR spectra of the products (for 8 at δ = 159.5 and 161.5 ppm for 9) and IR spectra (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 1715 cm−1 for 8).
O at 1715 cm−1 for 8).
 | ||
| Scheme 3 Synthesis of urea chlorindiol adducts 8 and 9, and the outcomes of the oxidative diol cleavage. Reaction conditions: (i) 20 equiv. urea derivative, pyridine, Δ (85% yield for 8; 54% yield for 9); (ii) 1–1.7 equiv. Pb(OAc)4, THF, (Et3N for preparation of 10), r.t., (69% yield for 10; 71% yield for 11). | ||
 | ||
| Fig. 1 UV-vis spectra (CH2Cl2) of the compounds indicated. | ||
We previously reported multiple methods for the oxidative cleavage of the pyrroline β,β′-bond of dihydroxychlorins.15 Thus, treatment of the polar magenta diol 8 under classic diol cleavage reaction conditions (Pb(OAc)4)15b,16 resulted in the formation of a red non-polar compound in good yields (69%). Its porphyrin-like UV-vis spectrum (Fig. 1B), NMR spectra, and its composition as determined by HR-MS identified it to be the known porpholactam 10.17 Once again, the expulsion of smaller fragments from the putative medium-ring derivative establishing a stable ‘tetrapyrrolic’ architecture thwarted the formation of the target medium-sized ring-expanded compound. On the bright side, this 3-step pathway toward 10 is more convenient and significantly higher yielding (overall 29% from diol 2 at a 50 mg scale) than the previously described 3-step synthesis (∼13%).
While we did not know the fragmentation mechanism of the putative intermediate resulting from the oxidative cleavage of 8, we suspected that the replacement of the amide protons might hinder the fragmentation by, for instance, blocking amide-iminol-type equilibria. Indeed, oxidative diol cleavage of dimethylurea adduct 9 generated a compound in good yields possessing a composition of two hydrogen atoms less than the starting material (as per HR-MS).‡ Moreover, it was characterized by a significantly red-shifted chlorin-type optical spectrum (Fig. 1B). The general red-shift of the spectrum and particularly the much reduced extinction coefficient of its Soret band observed suggest the increase in non-planarity of the chromophore, while its general broadening suggest an increase of conformational flexibility.18 Its NMR spectra retained the two-fold symmetry of the starting material with an upfield shift of the inner protons (δ = 1.88 ppm) suggestive of a non-planar chromophore, and the presence of two lactam carbon atoms in different environments (δ = 173.1 and 155.1 ppm). Thus all spectroscopic evidence point toward the successful formation of the target expanded pyrrole-modified porphyrin 11.
A single crystal X-ray structure analysis of 11 provided the final proof for the unique connectivity of this porphyrinoid (Fig. 2). The pyrroline β,β′-bond was, as designed, oxidatively cleaved and both β-carbons were converted in this process to lactams that are incorporated into an 1,3,6-triazocine-2,4,8-trione ring that resulted from the fusion of the three annulated dimethylurea atoms with the five pyrroline atoms into a single 8-membered ring. The non-pyrrolic moiety assumes a significantly non-planar conformation with an almost 90° twist along its long axis. This twist translates into the porphyrinic framework, leading to a mildly ruffled conformation.
 | ||
| Fig. 2 Stick representation of the single crystal X-ray structure of 11.§ All disorder, solvent molecules, and sp2-CH hydrogens were omitted for clarity. For details, see ESI.† | ||
The structure rationalizes all spectroscopic findings, but it also raises the question whether a larger than 8-membered ring will assume a figure-eight conformation that then will have a much weaker conformational effect on the remainder of the chromophore. This might rationalize why, for example, pyrrole-expanded porphyrazine 6 possesses a surprisingly regular chlorin-like optical spectrum (its solid state structure was not reported).10
In summary, the ‘mending and breaking’ strategy allowed firstly the incorporation of an 8-membered heterocycle into a pyrrole-modified porphyrin. The use of N-methylated derivatives was crucial in preventing the fragmentation of the newly formed heterocycle to generate a planar porphyrinoid containing four five-membered rings. Future systematic studies on larger ring systems now likely accessible using the ‘mending and breaking’ strategy while being mindful about blocking possible macrocycle degradation pathways will illuminate the relationship between ring-size of the non-pyrrolic building block and the optical properties of the chromophore.
Support through NSF grants CHE-1058846 and CHE-1465133 (to CB) is gratefully acknowledged.
Notes and references
- M. Stepien and L. Latos-Grazynski, Top. Heterocycl. Chem., 2009, 19, 83–153 CrossRef CAS.
- (a) J. L. Sessler and J. M. Davis, Acc. Chem. Res., 2001, 34, 989–997 CrossRef CAS PubMed; (b) A. Cetin and C. J. Ziegler, Dalton Trans., 2005, 25–26 RSC.
- M. Toganoh and H. Furuta, Chem. Commun., 2012, 48, 937–954 RSC.
- (a) P. J. Chmielewski and L. Latos-Grazynski, Coord. Chem. Rev., 2005, 249, 2510–2533 CrossRef CAS; (b) I. Gupta and M. Ravikanth, Coord. Chem. Rev., 2006, 250, 468–518 CrossRef CAS; (c) L. Arnold and K. Müllen, J. Porphyrins Phthalocyanines, 2011, 15, 757–779 CrossRef CAS; (d) T. D. Lash, J. Porphyrins Phthalocyanines, 2012, 15, 423–433 CrossRef; (e) C. Brückner, J. Akhigbe and L. Samankumara, in Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific, River Edge, NY, 2014, vol. 31, pp. 1–276 Search PubMed; (f) L. D. Costa, J. I. Costa and A. C. Tome, Molecules, 2016, 21 Search PubMed; (g) T. D. Lash, Acc. Chem. Res., 2016, 49, 471–482 CrossRef CAS PubMed.
- C. Brückner, Acc. Chem. Res., 2016, 49, 1080–1092 CrossRef PubMed.
- C. Brückner, D. C. G. Götz, S. P. Fox, C. Ryppa, J. R. McCarthy, T. Bruhn, J. Akhigbe, S. Banerjee, P. Daddario, H. W. Daniell, M. Zeller, R. W. Boyle and G. Bringmann, J. Am. Chem. Soc., 2011, 133, 8740–8752 CrossRef PubMed.
- (a) J. Akhigbe, G. Peters, M. Zeller and C. Brückner, Org. Biomol. Chem., 2011, 9, 2306–2313 RSC; (b) J. Akhigbe, L. Samankumara and C. Brückner, Tetrahedron Lett., 2012, 53, 3524–3526 CrossRef CAS.
- S. Shimizu, H. Zhu and N. Kobayashi, Chem. Commun., 2011, 47, 3072–3074 RSC.
- L. Yet, Chem. Rev., 2000, 100, 2963–3007 CrossRef CAS PubMed.
- A. G. Montalban, S. M. Baum, A. G. M. Barrett and B. M. Hoffman, Dalton Trans., 2003, 2093–2102 RSC.
- (a) H. W. Daniell, S. C. Williams, H. A. Jenkins and C. Brückner, Tetrahedron Lett., 2003, 44, 4045–4049 CrossRef CAS; (b) M. J. Crossley, P. L. Burn, S. J. Langford, S. M. Pyke and A. G. Stark, J. Chem. Soc., Chem. Commun., 1991, 1567–1568 RSC.
- J. Akhigbe and C. Brückner, Eur. J. Org. Chem., 2013, 3876–3884 CrossRef CAS.
- N. S. Hush, J. R. Reimers, L. E. Hall, L. A. Johnston and M. J. Crossley, Ann. N. Y. Acad. Sci., 1998, 852, 1–21 CrossRef CAS.
- C. Brückner, J. R. McCarthy, H. W. Daniell, Z. D. Pendon, R. P. Ilagan, T. M. Francis, L. Ren, R. R. Birge and H. A. Frank, Chem. Phys., 2003, 294, 285–303 CrossRef.
- (a) J. Akhigbe, C. Ryppa, M. Zeller and C. Brückner, J. Org. Chem., 2009, 74, 4927–4933 CrossRef CAS PubMed; (b) C. Brückner, S. J. Rettig and D. Dolphin, J. Org. Chem., 1998, 63, 2094–2098 CrossRef; (c) S. Banerjee, M. Zeller and C. Brückner, J. Porphyrins Phthalocyanines, 2012, 16, 576–588 CrossRef CAS; (d) J. R. McCarthy, H. A. Jenkins and C. Brückner, Org. Lett., 2003, 5, 19–22 CrossRef CAS PubMed.
- K. R. Adams, R. Bonnett, P. J. Burke, A. Salgado and M. A. Valles, J. Chem. Soc., Chem. Commun., 1993, 1860–1861 RSC.
- J. Akhigbe, J. P. Haskoor, J. A. Krause, M. Zeller and C. Brückner, Org. Biomol. Chem., 2013, 11, 3616–3628 CAS.
- H. Ryeng and A. Ghosh, J. Am. Chem. Soc., 2002, 124, 8099–8103 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Full description of the experimental details and a reproduction of key spectroscopic data of the novel compounds are provided. CCDC 1479383. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc04028d |
‡ Select spectral data for 8: 1H NMR (400 MHz, CDCl3): δ 8.64 (d, 3J = 4.7 Hz, H), 8.49 (s, 1H), 8.21 (s, 1H), 8.14 (d, 3J = 5.8 Hz, 3H), 7.86 (d, 3J = 6.8 Hz, 1H), 7.72 (m, 6H), 5.34 (br s, 1H, exchangeable with D2O), 4.26 (br s, 1H, exchangeable with D2O), −2.03 (br s, 1H, exchangeable with D2O) ppm; UV-vis (CH2Cl2) λmax (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ε) 406 (5.41), 512 (4.30), 541 (4.30), 591 (3.96), 642 (4.50) nm; HR-MS (ESI+, 100% CH3CN, TOF) m/z cald for C45H33N6O3 ([M·H]+), 705.2609, found 705.2605. For 9: 1H NMR (400 MHz, CDCl3): δ 8.57 (d, 3J = 4.9 Hz, 1H), 8.45 (s, 1H), 8.16 (d, 3J = 6.7 Hz, 1H), 8.07 (d, 3J = 5.2 Hz, 4H), 7.78–7.67 (m, 6H), 4.55 (s, 1H, exchangeable with D2O), 2.28 (s, 3H), −1.75 (s, 1H, exchangeable with D2O) ppm; UV-vis (CH2Cl2) λmax (logε) 410 (4.99), 520 (3.84), 549 (3.88), 596 (3.53), 649 (4.08) nm; HR-MS (ESI−, 100% CH3CN, TOF) m/z cald for C47H37N6O3 ([M·H]+) 733.2927, found 733.2941. For 11: 1H NMR (400 MHz, CDCl3): δ 8.35 (d, 3J = 4.9 Hz, 1H), 8.28 (br s, 1H), 8.16 (s, 1H), 8.04 (d, 3J = 4.9 Hz, 1H), 7.99 (br s, 2H), 7.70 (m, 4H), 7.56 (t, 3J = 7.5 Hz, 1H), 7.43 (br s, 1H), 7.10 (br s, 1H), 3.28–3.15 (s, 3H), 1.88 (s, 1H, exchangeable with D2O) ppm; UV-vis (CH2Cl2) λmax (logε) 455 (3.67), 585 (2.44), 625 (2.45), 740 (2.48) nm; HR-MS (ESI+, 100% CH3CN, TOF) m/z cald for C47H34O6N3 ([M·H]+) 731.2771, found 731.2775. For full spectroscopic characterization, see ESI.† ε) 406 (5.41), 512 (4.30), 541 (4.30), 591 (3.96), 642 (4.50) nm; HR-MS (ESI+, 100% CH3CN, TOF) m/z cald for C45H33N6O3 ([M·H]+), 705.2609, found 705.2605. For 9: 1H NMR (400 MHz, CDCl3): δ 8.57 (d, 3J = 4.9 Hz, 1H), 8.45 (s, 1H), 8.16 (d, 3J = 6.7 Hz, 1H), 8.07 (d, 3J = 5.2 Hz, 4H), 7.78–7.67 (m, 6H), 4.55 (s, 1H, exchangeable with D2O), 2.28 (s, 3H), −1.75 (s, 1H, exchangeable with D2O) ppm; UV-vis (CH2Cl2) λmax (logε) 410 (4.99), 520 (3.84), 549 (3.88), 596 (3.53), 649 (4.08) nm; HR-MS (ESI−, 100% CH3CN, TOF) m/z cald for C47H37N6O3 ([M·H]+) 733.2927, found 733.2941. For 11: 1H NMR (400 MHz, CDCl3): δ 8.35 (d, 3J = 4.9 Hz, 1H), 8.28 (br s, 1H), 8.16 (s, 1H), 8.04 (d, 3J = 4.9 Hz, 1H), 7.99 (br s, 2H), 7.70 (m, 4H), 7.56 (t, 3J = 7.5 Hz, 1H), 7.43 (br s, 1H), 7.10 (br s, 1H), 3.28–3.15 (s, 3H), 1.88 (s, 1H, exchangeable with D2O) ppm; UV-vis (CH2Cl2) λmax (logε) 455 (3.67), 585 (2.44), 625 (2.45), 740 (2.48) nm; HR-MS (ESI+, 100% CH3CN, TOF) m/z cald for C47H34O6N3 ([M·H]+) 731.2771, found 731.2775. For full spectroscopic characterization, see ESI.† |
| § Select crystallographic data for 11: C47H34N6O3, M = 730.80, orthorhombic, a = 26.853(2) Å, b = 10.1988(10) Å, c = 27.005(3) Å, V = 7395.8(13) Å3, T = 100 K, space group Pca21 (no. 29), Z = 8, 68142 reflections measured, 11219 unique (Rint = 0.1397), which were used in all calculations. The final R value was 0.1570 (R[F2 > 2σ(F2)]). CCDC 1479383. See ESI,† for the full description of the X-ray diffraction analysis of 11. |
| This journal is © The Royal Society of Chemistry 2016 |