
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFully reversible three-state interconversion of metallosupramolecular architectures†
Nikita
Mittal‡
,
Manik Lal
Saha‡
and
Michael
Schmittel
*
Center of Micro and Nanochemistry and Engineering, Organische Chemie I, Universität Siegen, Adolf-Reichwein-Str. 2, D-57068 Siegen, Germany. E-mail: schmittel@chemie.uni-siegen.de
First published on 24th June 2016
Abstract
The reversible switching of a sterically encumbered phenanthroline–Cu+–picolinaldehyde trio back and forth between homoleptic and heteroleptic coordination using the relative metal-ion to ligand ratio is the basis for an unprecedented cyclic three-state interconversion of metallacycles.
Stimuli-responsive structural rearrangements of supramolecular architectures1 are rapidly gaining popularity as a method to drive functional nanodevices and nanomachines.2 In this context, the notion of clean supramolecule-to-supramolecule transformations has opened a multifaceted venue to spectacular two-state switching systems, mostly based on the (electro)-/(photo)chemical bistability of metallacycles or cages.3 While a variety of stimuli (chemicals,4–6 redox,7 light,8 concentration9) have been applied successfully, further improvements are needed with regard to full and quantitative reversibility, acceleration of transformations as well as access to multi-state and non-equilibrium systems.10
Multi-state transformations with three or more well-defined supramolecular states should be useful for creating molecular devices and logic gates of higher complexity.11 Such transformations, though, have rarely been explored,12 because they require multiple rearrangements of (metallo)supramolecular architectures, with each of the individual states representing a thermodynamic minimum protected by a sufficient energy barrier. Ideally, this barrier should vanish upon addition of the stimulus. Nitschke and coworkers have recently reported the cascading network of transformations of iminopyridine metal complexes, using subcomponent substitution reactions.12 While these rearrangements are complex and unique, they represent irreversible transformations preventing their use in reversible switching processes. A three-state reversible transformation demands a design with a cyclic interconversion of the involved (metallo)supramolecules. To this end, we herein demonstrate the unprecedented and quantitative three-state cyclic interconversion of three metallacycles, namely of the square S, the isosceles triangle T and the rectangle R, based on the reversible switching of a sterically encumbered phenanthroline–copper(I)–picolinaldehyde trio back and forth between homoleptic and heteroleptic coordination (Scheme 1).
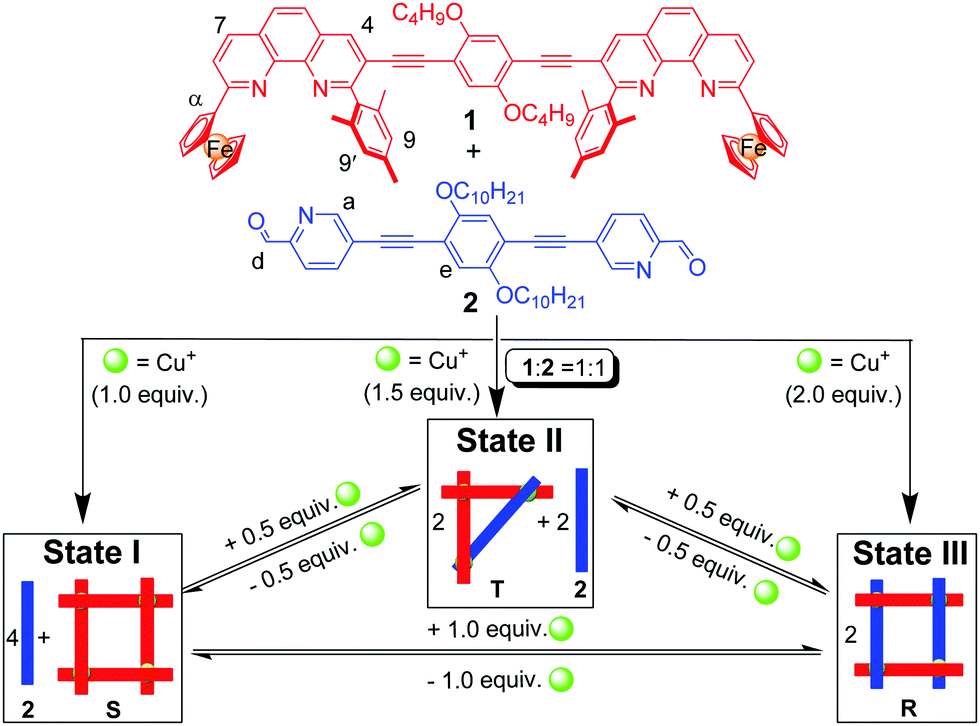 | ||
| Scheme 1 Three-state cyclic interconversion of the metallosupramolecular complexes S, T and R depending on the Cu+ amount (equiv. refer to the amount of 1). | ||
Conceptually, the overall processes of Scheme 1 can be viewed as three-state supramolecular transformations driven by completive vs. incomplete self-sorting protocols.13 It requires the careful design of two ditopic ligands L1, L2 and a metal ion Mn+ to afford three distinct states depending on the L1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :L2:Mn+ ratio. The states involve self-sorting into a heteroleptic structure, e.g. [M4(L1)2(L2)2]4n+ (completive self-sorting), at L1:L2:Mn+ = 1:1:2 and an entirely homoleptic assembly in presence of the other ligand, e.g. [M4(L1)4]4n+ + 4 × L2 (incomplete self-sorting), at L1:L2:Mn+ = 1:1:1. In addition, a third state, i.e. an architecture encompassing both homoleptic and heteroleptic coordination motifs, is required at an intermediate metal ion's stoichiometry, again in an incomplete self-sorting (see Scheme 1, state II). The structures R, S and T have been chosen, because of their small number of metal complexation units, which should make (inter)conversions kinetically feasible.
:L2:Mn+ ratio. The states involve self-sorting into a heteroleptic structure, e.g. [M4(L1)2(L2)2]4n+ (completive self-sorting), at L1:L2:Mn+ = 1:1:2 and an entirely homoleptic assembly in presence of the other ligand, e.g. [M4(L1)4]4n+ + 4 × L2 (incomplete self-sorting), at L1:L2:Mn+ = 1:1:1. In addition, a third state, i.e. an architecture encompassing both homoleptic and heteroleptic coordination motifs, is required at an intermediate metal ion's stoichiometry, again in an incomplete self-sorting (see Scheme 1, state II). The structures R, S and T have been chosen, because of their small number of metal complexation units, which should make (inter)conversions kinetically feasible.
To identify real binding motifs in L1 and L2, one has to consider the binding constants in combination with maximum site occupancy.14 For state I (Scheme 1), the binding constants of L1 = 1 and L2 = 2 should follow the order of logK ([M(L1)2]n+) ≫ logK ([M(L2)2]n+), while the heteroleptic complex [M(L1)(L2)]n+ required in state III should be much more stable than the two homoleptic counterparts in equilibrium. To realise the three-state shuffling, the 2-ferrocenyl-9-mesityl-[1,10]-phenanthroline (3) (Chart 1) was selected as a prototype ligand in L1 (vide supra) and tested first at the mononuclear level (Scheme 2). As established recently,15 the encumbered ligand 3 still allows clean formation of its homoleptic complex C1 = [Cu(3)2]PF6, but due to its strong electron-donating character might also cleanly furnish heteroleptic complexes with the appropriate counterparts. Ligand 3 works with both steric and donor effects, thus different from ligands in HETPHEN (heteroleptic bisphenanthroline16) complexes relying solely on steric factors.
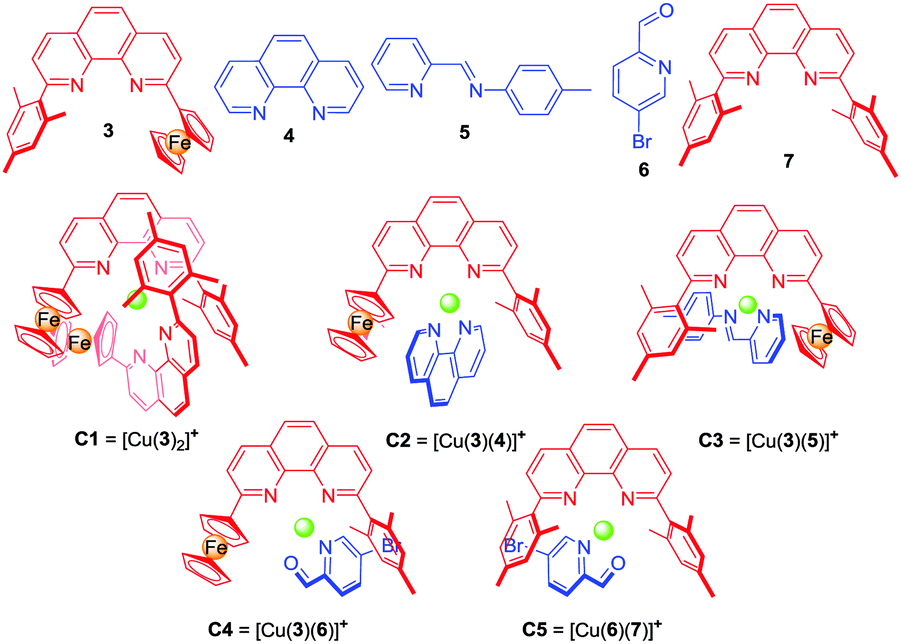 | ||
| Chart 1 Ligands 3–7 and model complexes C1–C5. | ||
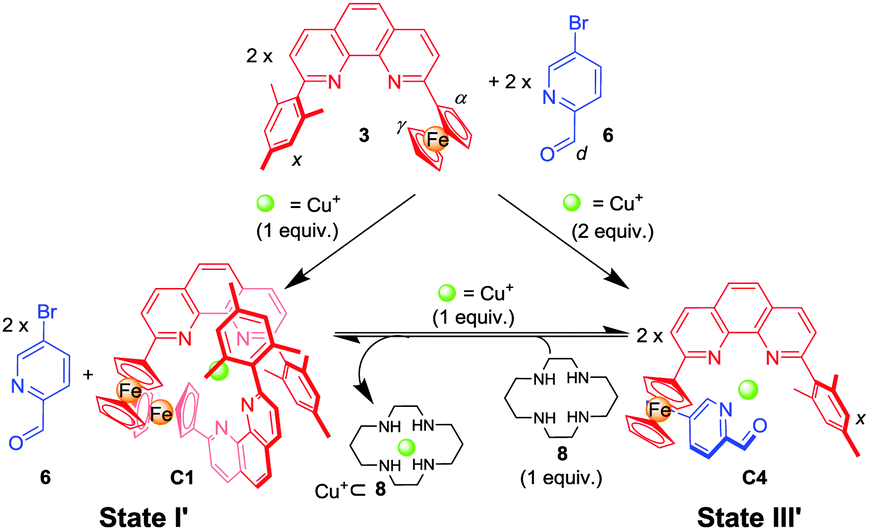 | ||
| Scheme 2 Stoichiometry-controlled switching between mononuclear complexes C1 and C4 (all equiv. related to ligand 3). | ||
Unfortunately, the heteroleptic complex C2 = [Cu(3)(4)]PF6 did not form in quantitative yield from an equimolar mixture of 3, 4 and [Cu(CH3CN)4]PF6 (Fig. S3, ESI†). Apparently, the stability of the corresponding pair of homoleptic complexes C1 = [Cu(3)2]PF6 and [Cu(4)2]PF6 is partly competitive to that of the desired heteroleptic complex C2 = [Cu(3)(4)]PF6.
To realise a heteroleptic copper complex [Cu(3)(L)]+ as demanded in state III′ (Scheme 2), we envisaged to utilise weaker donor ligands L, such as 4-methyl-N-(pyridin-2-ylmethylene)aniline (5)17 or picolinaldehyde 6 as counterparts. The reaction of a 1:1:1 mixture of 3, 5 and Cu+ furnished the desired complex C3 = [Cu(3)(5)]PF6, however, still contaminated with traces of homoleptic complexes (Fig. S5, ESI†), thus suggesting a further reduction of the donor strength of L. We finally turned our attention to picolinaldehyde 616b,18 and, indeed, a 1:1:1 mixture of 3, 6 and Cu+ ions furnished the heteroleptic complex C4 = [Cu(3)(6)]PF6 in a clean manner (Fig. S7, S8 and S30, ESI†). The 1H NMR showed a diagnostic upfield shift of the aldehyde proton (d-H) in C4 (δ = 9.85 ppm), which is similar to that in C5 (δ = 9.63 ppm)18a and different from that in the free ligand 6 (δ = 10.08 ppm), due to the intimate π–π stacking(s) between the mesityl/ferrocenyl groups of 3 and the pyridine-2-carbaldehyde unit of 6.
Now it was time to test whether 6 can play the desired role as an innocent bystander in state I′ (Scheme 2). A 2:2:1 mixture of 3, 6 and Cu+ ions afforded in an incomplete self-sorting the required state I′ = C1 + 6 as shown by NMR data (Fig. S9 and S10, ESI†). Using ligand 6 and the complexes C115 and C4 allowed unambiguous assignment of the 1H-NMR data in state I′.
State II in Scheme 1 requires orthogonality of C1 and C4.17a Upon mixing Cu+ ions, ligands 3 and 6 in a 2:3:1 ratio, the 1H NMR spectra of the resulting mixture showed that both C1 and C4 emerged in a 1:1 ratio, thereby confirming their orthogonality (Fig. S12, ESI†).
Subsequently, the reversibility of the interconversion of C1 + 2 × 6 ⇌ 2 × C4 was interrogated as a function of the metal stoichiometry (Fig. 1). Addition of 1 equiv. of Cu+ ions (with respect to the initial amount of ligand 3) to the solution of C1 + 2 × 6 produced a 1H NMR spectrum that precisely matched that of C4. When 1 equiv. of cyclam (8) (with respect to the initial amount of ligand 3) was added to the resultant solution of C4, the initial (C1 + 2 × 6) state was recovered. We performed this reversible switching over three cycles and found that the transformations were completely reversible (see Fig. S11, ESI†).
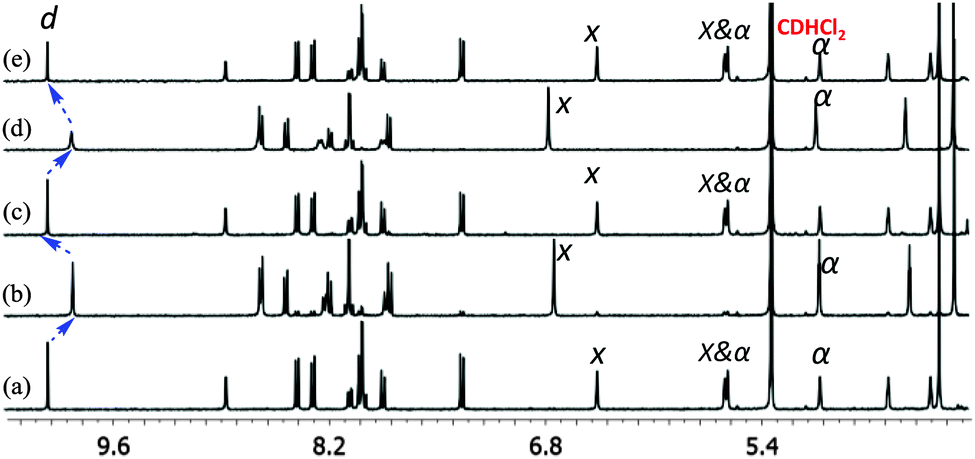 | ||
| Fig. 1 Partial 1H NMR spectra (400 MHz, CD2Cl2, 298 K) depicting the reversible switching cycles between (C1 + 2 × 6) and (2 × C4). (a), (c) and (e) correspond to (C1 + 2 × 6) whereas (b) and (d) represent C4, obtained after successive addition of Cu+ (1 eq.) and cyclam (1 eq.). Some selected protons are assigned. | ||
To test the three-state transformation of Scheme 1, the new ligand 1 was synthesised by means of a palladium catalyzed Sonogashira coupling reaction between 1,4-bis(decyloxy)-2,5-diiodobenzene and 3-ethynyl-2-mesityl-9-ferrocenyl-[1,10]-phenanthroline (see ESI†), both known compounds.15 Subsequently, we prepared the supramolecular structures S, T and R (Chart 2) as references for the stoichiometry-controlled shuffling (Scheme 1 and Fig. 2). At first, ligand 1 and [Cu(CH3CN)4]PF6 were mixed in a 1:1 ratio and refluxed for 30 min in DCM. The resultant clear red complex was characterised by ESI-MS, 1H-NMR, diffusion-ordered spectroscopy (DOSY) and by elemental analysis. The ESI-MS spectrum (Fig. S32, ESI†) exhibited only two peaks at 1294.5 Da and 1774.3 Da in the region of 200–2000 Da representing S = [Cu4(1)4](PF6)n(4−n)+ (n = 0 and 1). Therefore, the ESI-MS data suggest that square S is the sole product of the reaction, which was further ascertained by the DOSY NMR (see Fig. S25, ESI†) showing a single species with a diffusion coefficient of 3.4 × 10−10 m2 s−1. The structure of S was also confirmed by the expected 1H-NMR pattern because the ferrocenyl α-H protons of 1 were split into two peaks due to their diastereotopicity in the C1-type of corners (see Fig. S13 and S14, ESI†). Owing to the four stereogenic axes in assembly S, various diastereomers are present in the 1H-NMR.
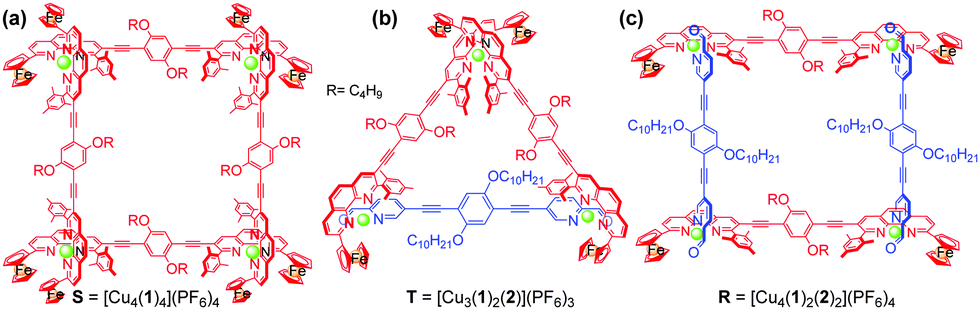 | ||
| Chart 2 Chemical structures of (a) square, (b) triangle and (c) rectangle. | ||
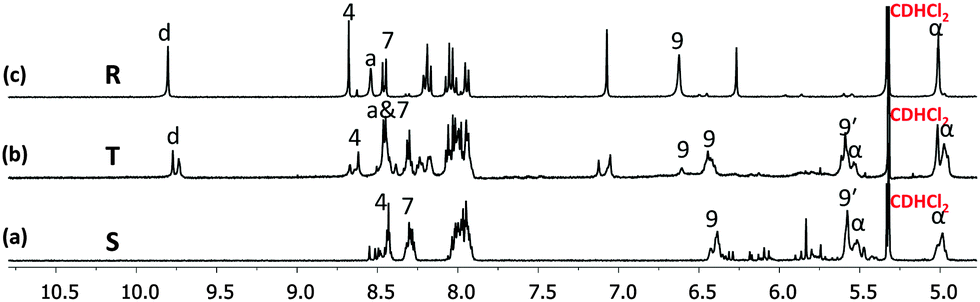 | ||
| Fig. 2 Partial 1H NMR spectra (400 MHz, CD2Cl2, 298 K) of (a) square S, (b) triangle T and (c) rectangle R. Selected protons are assigned. | ||
Similarly, the self-assembly of [Cu(CH3CN)4]PF6 and ligands 1 & 2 = 2:1:1 produced the rectangle R = [Cu4(1)2(2)2](PF6)4 as the sole product. A single diffusion coefficient in the DOSY NMR (D = 4.5 × 10−10 m2 s−1) as well as a single set of signals in the 1H-NMR provided evidence for its purity (Fig. S27 and S17, ESI†). As seen in the 1H NMR, the aldehyde protons d-H of 2 in the C4-type complex unit in R appear at 9.80 ppm, thus significantly shifted upfield by 0.26 ppm compared with the free ligand 2. Similar to model complex C4, the 1H-NMR of rectangle R exhibited only one type of ferrocenyl α-H, which further confirms the heteroleptic complexation motif (Fig. S18, ESI†). The ESI-MS spectrum exhibited two peaks at m/z = 1003.5 and 1385.6 Da for [Cu4(1)2(2)2](PF6)n(4−n)+ for n = 0 and 1, respectively, that clearly support the integrity of R (see Fig. S34, ESI†).
Finally, the isosceles triangle T was prepared from a mixture of 1, 2 and [Cu(CH3CN)4]PF6 = 2:1:3 ratio after reflux for 1 h in acetonitrile/DCM (1:3). The ESI-MS spectrum shows a single major peak at 1722.7 Da for T = [Cu(1)2(2)](PF6)2+ which suggests quantitative formation (see Fig. S33, ESI†). The formation of the isosceles triangle T was then further confirmed by 1H-NMR, 1H–1H COSY NMR as well as with DOSY NMR, the latter displaying a single entity at D = 4.3 × 10−10 m2 s−1 (Fig. S26, ESI†). In the 1H-NMR, the aldehyde protons d-H of ligand 2 is diagnostically shifted from 10.09 to 9.77 & 9.74 ppm while the splitting of the ferrocenyl α-H equally suggests the presence of both homoleptic C1-type and heteroleptic C4-type corners (Fig. S15 and S16, ESI†).
The clean formation of S, R and T provides a reliable base for investigating the incomplete vs. completive self-sorting events using a varying copper(I) stoichiometry. Thus, in order to verify the shuffling as suggested in Scheme 1, first ligands 1 and 2 as well as [Cu(CH3CN)4]PF6 were mixed in 1:1:1 ratio in CH2Cl2/CH3CN to attain state I (Scheme 1). Indeed, the 1H NMR of the mixture confirmed the formation of square S = [Cu4(1)4]4+ while ligand 2 remained free in solution (Fig. 3a). In the 1H NMR the diagnostic ferrocenyl protons of ligand 1 (α-H) split into two peaks identical to that for independently prepared square S, while the characteristic aldehyde proton d-H of 2 appears at the same position as in the free ligand 2 (see Fig. S20, ESI†). The DOSY NMR showed two compounds with well distinguishable diffusion coefficients, i.e. 3.3 × 10−10 m2 s−1 (S) and 7.9 × 10−10 m2 s−1 (2), in the mixture (see Fig. S28, ESI†), which confirmed the presence of state I in the mixture.
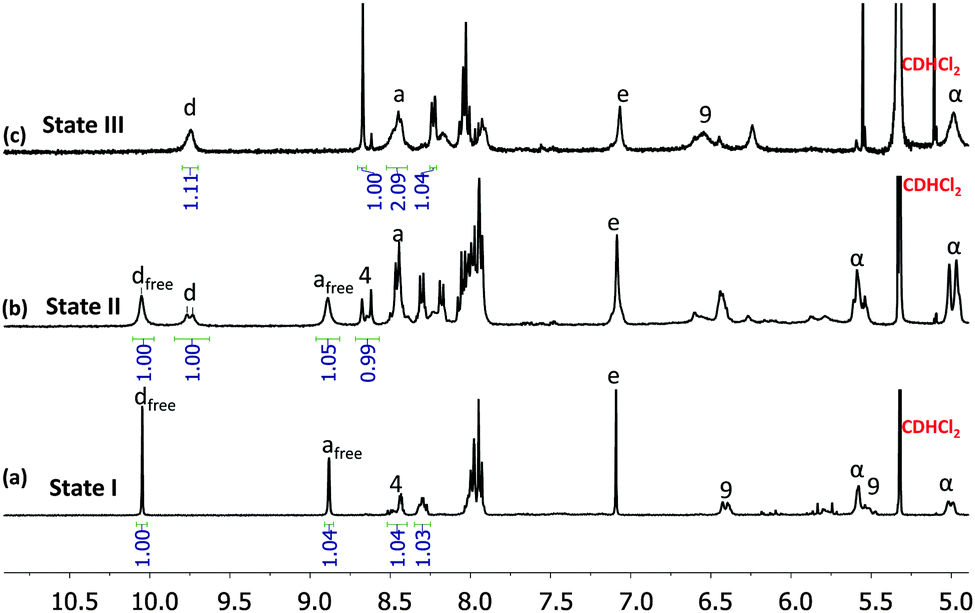 | ||
| Fig. 3 Partial 1H NMR spectra for comparison (400 MHz, CD2Cl2, 298 K) of ligand 1 and ligand 2 in 1:1 ratio (a) with 1 eq. Cu+; (b) with 1.5 eq. Cu+; (c) with 2 eq. Cu+. Some selected protons are assigned. | ||
We then added 0.5 equiv. of [Cu(CH3CN)4]PF6 (relative to 1) to the above solution and refluxed it for 1 h to generate state II, i.e. triangle T and free ligand 2 (0.5 equiv.). The 1H NMR showed two sets of aldehyde protons d-H at a 1:1 ratio suggesting two distinct CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups, one representing the triangle T = [Cu3(1)2(2)](PF6)3 and the other the free ligand 2 (Fig. 3b), as expected for state II with its incomplete self-sorting. Comparison of the 1H NMR (see Fig. S22, ESI†) of state II with that of the independently prepared triangle T and ligand 2 confirmed the above assignment. The DOSY NMR (see Fig. S29, ESI†) of state II diagnostically showed two different species with D = 4.2 × 10−10 and 6.9 × 10−10 m2 s−1 that were assigned to triangle T and ligand 2, respectively. The ESI-MS (Fig. S35, ESI†) also showed one major peak of T and one minor peak of ligand 2 corroborating state II.
O groups, one representing the triangle T = [Cu3(1)2(2)](PF6)3 and the other the free ligand 2 (Fig. 3b), as expected for state II with its incomplete self-sorting. Comparison of the 1H NMR (see Fig. S22, ESI†) of state II with that of the independently prepared triangle T and ligand 2 confirmed the above assignment. The DOSY NMR (see Fig. S29, ESI†) of state II diagnostically showed two different species with D = 4.2 × 10−10 and 6.9 × 10−10 m2 s−1 that were assigned to triangle T and ligand 2, respectively. The ESI-MS (Fig. S35, ESI†) also showed one major peak of T and one minor peak of ligand 2 corroborating state II.
To conclude the three-state reshuffling, we added a further 0.5 equiv. of Cu+ to the above solution and characterised it by 1H NMR, ESI-MS after heating for 2 h. The 1H NMR of the resultant mixture (Fig. 3c) is basically identical with the 1H NMR of the pure rectangle R, which directly confirmed the presence of state III in the mixture.
After attaining all states of Scheme 1 in a single solution by controlling the Cu+ ion stoichiometry, we finally tested the reversibility of this system by removing and adding Cu+ ion. We used cyclam to remove Cu+ ion as in our model system (Scheme 2). Confirming our design, the system turned out to be fully reversible without any loss. For example state III was easily converted into state II by just adding 0.5 equiv. of cyclam (see Fig. S24d, ESI†). Similarly states II and state III were fully switched into state I by addition of 0.5 and 1 equiv. of cyclam, respectively (see Fig. S24, ESI†). Finally, state I was converted into state II or III or sequentially from state I to state II to state III by controlling the metal ion stoichiometry (see Fig. S23 and S24, ESI†).
The MM+ force-field calculations on square S, isosceles triangle T and rectangle R allow the modeling of these supramolecular architectures (see Fig. S37–S39, ESI†) and provided some insight about their structures. Taking the metal–metal distances in the energy minimised structure as a measure, the four metal corners are separated by 1.83 nm each in square S, in isosceles triangle three metal corners are distant by 1.80, 1.80 and 1.77 nm and again four metal corners in rectangle R are separated by 1.82, 1.72, 1.82, 1.72 nm.
In conclusion, we present the fully interconvertible and reversible cyclic transformation of three metallosupramolecular architectures, i.e. square S, triangle T and rectangle R. The clean and quantitative (inter)conversion of one structure into another one is the result of delicate self-sorting preferences allowing full control over homoleptic vs. heteroleptic vs. mixed homo- & heteroleptic complexation depending on metal addition/removal. To our knowledge this cycle is the first example of a fully reversible three-state transformation of supramolecular structures by varying the metal-ion:ligand ratio.
We thank the DFG (Schm 647/20-1) and the University of Siegen for continued support.
Notes and references
- (a) J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151 RSC; (b) A. J. McConnell, C. S. Wood, P. P. Neelakandan and J. R. Nitschke, Chem. Rev., 2015, 115, 7729 CrossRef CAS PubMed.
- (a) A. Coskun, M. Banaszak, R. D. Astumian, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2012, 41, 19 RSC; (b) U. Lüning, Angew. Chem., Int. Ed., 2012, 51, 8163 CrossRef PubMed.
- (a) T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001 CrossRef CAS PubMed; (b) W. Wang, Y.-X. Wang and H.-B. Yang, Chem. Soc. Rev., 2016, 45, 2656 RSC.
- Metal ion and/or ligand: (a) K. Harano, S. Hiraoka and M. Shionoya, J. Am. Chem. Soc., 2007, 129, 5300 CrossRef CAS PubMed; (b) S. Hiraoka, Y. Sakata and M. Shionoya, J. Am. Chem. Soc., 2008, 130, 10058 CrossRef CAS PubMed; (c) J. E. Betancourt, M. Martín-Hidalgo, V. Gubala and J. M. Rivera, J. Am. Chem. Soc., 2009, 131, 3186 CrossRef CAS PubMed; (d) P. J. Lusby, P. Müller, S. J. Pike and A. M. Z. Slawin, J. Am. Chem. Soc., 2009, 131, 16398 CrossRef CAS PubMed; (e) Y.-R. Zheng, Z. Zhao, M. Wang, K. Ghosh, J. B. Pollock, T. R. Cook and P. J. Stang, J. Am. Chem. Soc., 2010, 132, 16873 CrossRef CAS PubMed; (f) M. L. Saha, S. Pramanik and M. Schmittel, Chem. Commun., 2012, 48, 9459 RSC; (g) M. L. Saha, K. Mahata, D. Samanta, V. Kalsani, J. Fan, J. W. Bats and M. Schmittel, Dalton Trans., 2013, 42, 12840 RSC; (h) S. Neogi, Y. Lorenz, M. Engeser, D. Samanta and M. Schmittel, Inorg. Chem., 2013, 52, 6975 CrossRef CAS PubMed; (i) O. Jurček, P. Bonakdarzadeh, E. Kalenius, J. M. Linnanto, M. Groessl, R. Knochenmuss, J. A. Ihalainen and K. Rissanen, Angew. Chem., Int. Ed., 2015, 54, 15462 CrossRef PubMed.
- Solvent: (a) J. Ramírez, A.-M. Stadler, N. Kyritsakas and J.-M. Lehn, Chem. Commun., 2007, 237 RSC; (b) B. Kilbas, S. Mirtschin, R. Scopelliti and K. Severin, Chem. Sci., 2012, 3, 701 RSC.
- Reagent: L. Zhao, B. H. Northrop and P. J. Stang, J. Am. Chem. Soc., 2008, 130, 11886 CrossRef CAS PubMed.
- Redox source: A.-M. Stadler, C. Burg, J. Ramírez and J.-M. Lehn, Chem. Commun., 2013, 49, 5733 RSC.
- Light: (a) S. Chen, L.-J. Chen, H.-B. Yang, H. Tian and W. Zhu, J. Am. Chem. Soc., 2012, 134, 13596 CrossRef CAS PubMed; (b) M. Han, R. Michel, B. He, Y.-S. Chen, D. Stalke, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2013, 52, 1319 CrossRef CAS PubMed; (c) M. Han, Y. Luo, B. Damaschke, L. Gómez, X. Ribas, A. Jose, P. Peretzki, M. Seibt and G. H. Clever, Angew. Chem., Int. Ed., 2016, 55, 445 CrossRef CAS PubMed.
- Concentration: X. Lu, X. Li, K. Guo, T.-Z. Xie, C. N. Moorefield, C. Wesdemiotis and G. R. Newkome, J. Am. Chem. Soc., 2014, 136, 18149 CrossRef CAS PubMed.
- C. S. Wood, C. Browne, D. M. Wood and J. R. Nitschke, ACS Cent. Sci., 2015, 1, 504 CrossRef CAS PubMed.
- (a) L. F. Lindoy, Nature, 1993, 364, 17 CrossRef; (b) A. P. de Silva and S. Uchiyama, Nat. Nanotechnol., 2007, 2, 399 CrossRef CAS PubMed.
- (a) V. E. Campbell, X. de Hatten, N. Delsuc, B. Kauffmann, I. Huc and J. R. Nitschke, Nat. Chem., 2010, 2, 684 CrossRef CAS PubMed; (b) W. Meng, T. K. Ronson, J. K. Clegg and J. R. Nitschke, Angew. Chem., Int. Ed., 2013, 52, 1017 CrossRef CAS PubMed.
- M. L. Saha and M. Schmittel, Org. Biomol. Chem., 2012, 10, 4651 Search PubMed.
- R. Krämer, J.-M. Lehn and A. Marquis-Rigault, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 5394 CrossRef.
- M. L. Saha, N. Mittal, J. W. Bats and M. Schmittel, Chem. Commun., 2014, 50, 12189 RSC.
- (a) M. Schmittel and A. Ganz, Chem. Commun., 1997, 999 RSC; (b) M. L. Saha, S. Neogi and M. Schmittel, Dalton Trans., 2014, 43, 3815 RSC.
- (a) M. L. Saha, S. De, S. Pramanik and M. Schmittel, Chem. Soc. Rev., 2013, 42, 6860 RSC; (b) Depending on the substituent in the 4-position of anilines, the association constant Kass of iminopyridines towards [Cu(phenAr2)]+ (phenAr2 = 2,9-diaryl-[1,10]-phenanthroline) varies from 104 to 106 M−1.
- (a) M. Schmittel, M. L. Saha and J. Fan, Org. Lett., 2011, 13, 3916 CrossRef CAS PubMed; (b) J. Fan, M. L. Saha, B. Song, H. Schönherr and M. Schmittel, J. Am. Chem. Soc., 2012, 134, 150 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectroscopic data are provided for all supramolecular assemblies, complexes and ligand 1. See DOI: 10.1039/c6cc03824g |
| ‡ Nikita Mittal and Manik Lal Saha contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |