
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel properties and potential applications of functional ligand-modified polyoxotitanate cages
Ning
Li
ab,
Peter D.
Matthews
ac,
He-Kuan
Luo
b and
Dominic S.
Wright
*a
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: dsw1000@cam.ac.uk
bInstitute of Materials Research and Engineering, Agency for Science Technology and Research, 2 Fusionopolis Way, #08-03, Innovis, Singapore 138634
cSchool of Chemistry, University of Manchester, Oxford Road, M13 9PL, UK
First published on 14th June 2016
Abstract
Functional ligand-modified polyoxotitanate (L-POT) cages of the general type [TixOy(OR)z(L)m] (OR = alkoxide, L = functional ligand) can be regarded as molecular fragments of surface-sensitized solid-state TiO2, and are of value as models for studying the interfacial charge and energy transfer between the bound functional ligands and a bulk semiconductor surface. These L-POTs have also had a marked impact in many other research fields, such as single-source precursors for TiO2 deposition, inorganic–organic hybrid material construction, photocatalysis, photoluminescence, asymmetric catalysis and gas adsorption. Their atomically well-defined structures provide the basis for the understanding of structure/property relationships and ultimately for the rational design of new cages targeting specific uses. This highlight focuses on recent advances in L-POTs research, with emphasis on their novel properties and potential applications.
 Ning Li | Ning Li completed his BEng degree at the National University of Singapore (2009–2013), and undertook a one-year research attachment in the Institute of Materials Research and Engineering A*STAR Singapore, before joining the Wright group as a PhD student in 2014. His current work is on polyoxometalate cage structures and related functional materials. |
 Peter D. Matthews | Peter D. Matthews studied for his MSc degree at the University of Cambridge (2008–2012) and completed his PhD (2012–2016) in the Wright group working on polyoxotitanate cages and heteroatom doped graphites. He has since moved to the University of Manchester to take up an EPSRC Doctoral Prize with Prof. Paul O'Brien. |
 He-Kuan Luo | He-Kuan Luo received his PhD in chemistry in 1996 from Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences. He then worked as an Engineer/Senior Engineer at the Polyolefins National Engineering Research Centre, Beijing Research Institute of Chemical Industry SINOPEC, China (1997–2000); as an Alexander von Humboldt Research Fellow at the Technical University Berlin, Germany (2001–2002); as a Research Fellow at Queen's University, Belfast, UK (2003–2004); and as a Senior Research Fellow/Research Scientist at the Institute of Chemical and Engineering Sciences A*STAR Singapore (2005–2011). From 2012, he has been a Research Scientist at the Institute of Materials Research and Engineering A*STAR Singapore. His current research interests are metal–ligand coordination materials, and functional materials from sustainable resources such as biomass and CO2. |
 Dominic S. Wright | Dominic S. Wright obtained his first degree at Strathclyde University (1982–1986) before moving to Cambridge University where he did his PhD under the late Dr Ron Snaith (1986–1989). After a research fellowship at Gonville and Caius College Cambridge (1989–1991), he was appointed to a lectureship at Cambridge and promoted to Reader in 2002 and promoted to a personal chair in Inorganic Chemistry in 2010. He is the author of around 290 academic papers. He was awarded the RSC Main Group Chemistry Award in 2012. |
1. Introduction
In recent years titania (TiO2) has attracted a great deal of attention as a highly stable and environmentally benign material,1 with a broad range of important applications spanning from the well-known white pigment in paint2 to its application as a semiconductor in state-of-the-art sensors3,4 and photonic devices.5,6 In particular, there is a growing interest in the use of TiO2 for solar energy harvesting owing to pressing future energy needs and associated environmental concerns involved with fossil fuels. As a widely used semiconductor, solid-state TiO2 and its various nano-structural forms have been thoroughly documented in the literature.1 Detailed understanding of the TiO2 surface chemistry at the atomic level is, however, still lacking, mainly due to the inherent difficulties in exploring dynamic processes at the TiO2 surface where chemical reactions occur (e.g., water splitting, organic pollutant degradation), and to the insolubility of TiO2 in organic solvents, preventing studies using solution-based analytical techniques.Using recently developed synthetic approaches, the successful synthesis of atomically well-defined polyoxotitanate (POT) cages has allowed chemists to gain new insights into molecular activation using TiO2.7–9 Resembling the fragments of bulk TiO2, POT cages of the type [TixOy(OR)z] (OR = alkoxide), consisting of TixOy inorganic titanium oxide cores encapsulated within an alkoxide ligand periphery, can be considered as models for studying the structural chemistry of bulk TiO2, including its crystal growth mechanism,10,11 the electronic and structural effects of heterometallic doping,12–21 and the influence of surface functional ligand modification.22–31 While their excellent solubility in common organic solvents allows POTs to be studied using various standard methods (e.g., NMR,32–34 mass spectrometry35), single-crystal X-ray diffraction is perhaps the preeminent tool for their characterisation (allowing unambiguous characterisation of structural features which can be related to that of bulk TiO2 itself).9
We and others have recently reviewed advances in the area of metal-doped cages (M-POTs), with the emphasis on their structures, photochemistry and applications.7,9 Apart from metal doping, another major area of interest has been the modification of these POT cages with functional ligands that impart additional structural diversity and chemical reactivity, giving them a broad range of new applications. In this highlight we focus specifically on recent developments in functional ligand-modified POT cages (L-POTs) of this type, as well as functional ligand-modified M-POT cages (L-M-POTs). To our knowledge this is the first review of this important and developing area of molecular and materials chemistry, which directly links their molecular structures with their novel properties and potential applications. The relationship between the various classes of polyoxotitanate cages which will be discussed is shown schematically in Fig. 1. Major themes of this highlight are their novel ligand-introduced properties and potential applications, such as single-source precursors for TiO2 deposition, nano-building blocks (NBBs) for inorganic–organic hybrid materials construction, photocatalysis, photochromism, photoluminescence, asymmetric catalysis and gas adsorption.
 | ||
| Fig. 1 Illustration of the relationship between polyoxotitanate (POT) cages, metal-doped POT cages (M-POT) and ligand-modified POT cages (L-POT), and an L-M-POT cage that possesses both metal-doping and ligand-modification. The blue cubes assembly represents the TixOy core, while the orange cubes are metal dopant ions. | ||
2. Properties and applications of L-POTs
2.1 Single-source precursors for TiO2 deposition
The presence of the readily hydrolysable OR-groups in POT cages of the type [TixOy(OR)z] or M-POT cages [TixOy(OR)zMnXm] (M = a transition metal or lanthanide ion, X = an anion such as halide) introduces the prospect of their use as single-source precursors for the deposition of TiO2 materials (Scheme 1).7,11 Hydrolysis of the ligand-functionalised L-POT cages of the type [TixOy(OR)z(L)m] (OR = alkoxide, L = functional ligand) is, however, often hindered by the presence of more robust ligand peripheries (L) in the majority of cases. The latter often survive intact in aqueous conditions without degradation and may require special treatment such as heating, autoclaving and addition of specific nucleation tuners for degradation to TiO2 to occur.36–38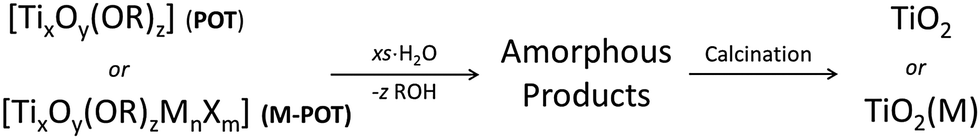 | ||
| Scheme 1 Reaction scheme of POT cages [TixOy(OR)z] or M-POT cages [TixOy(OR)zMnXm] upon hydrolysis and further calcination, forming TiO2 or metal-doped TiO2 [TiO2(M)]. The amorphous products initially formed probably consist of TiO2 mixed with organic residues. | ||
The titanium oxide directly generated from hydrolysis under various conditions is usually amorphous in nature, which upon calcinating at 400 °C is converted to crystalline anatase and further to rutile at 500 °C.39 The presence of functional ligands under these circumstances can allow the conversion to titania to follow a different reaction route. For example, the tetranuclear cage [Ti4(μ4-O)(μ-furfuryloxo)14] (1) can convert to TiO2via cationic polymerization using trifluoromethanesulfonic anhydride as the initiator (see Scheme 2).40 The phase purity of the TiO2 produced can be fine-tuned by altering the thermal treatment conditions. Pre-treatment with excess distilled water at 100 °C before calcination at 400 °C produces solely anatase, but directly calcinating at 400 °C gives rise to mixtures of anatase and rutile.40 Chemical vapour deposition (CVD) is another physical technique for mainly producing TiO2 thin films, using POTs/L-POTs as precursors thanks to their excellent solubility in organic carrier solvents.41 However, so far only mono- and di-nuclear titanium complexes have been reported as CVD precursors and the use of cages with higher nuclearity remains largely unexplored.42,43
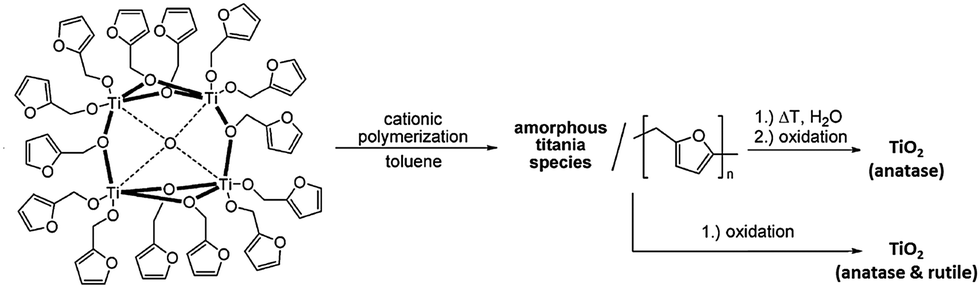 | ||
| Scheme 2 Reaction scheme for the cationic polymerization of the tetranuclear cage [Ti4(μ4-O)(μ-furfuryloxo)14] (1) into titania. Adapted with permission from ref. 40, ©2008 Wiley. | ||
More interestingly, self-assembled TiO2@C core–shell nanoparticles can be produced simply by heating the mononuclear complex [Ti(OEt)3(bdmap)] (Hbdmap = 1,3-bis-(dimethylamino)propanol) (2) in a N2-filled Swagelock cell at 700 °C.43 Hence, organic ligands do not always act as sources of detrimental carbon impurities for TiO2 crystallization, but instead can be employed to produce novel hybrid titania materials. This point has been further supported by other recent studies, in which the morphologies of the TiO2 nanostructures formed can be fine-tuned by varying the type of L-POT precursor employed and the reaction conditions.36,44
2.2 Nano-building blocks for hybrid materials construction
The nano-building block (NBB) based method is particularly attractive in synthesizing inorganic–organic hybrid materials, which combine the inorganic and organic components on the molecular scale to achieve a synergetic marriage of properties inherent to each constituent.45 As preformed monodispersed nano-objects that retain their molecular integrity and intrinsic properties during superstructure assembly, POT cages represent an attractive family of NBBs.46,47 Rozes and Sanchez8 have thoroughly reviewed the ‘Lego-Like’ construction of nanostructured hybrid materials using POT cages, including the sub-types of titanium-oxo-alkoxide [TinOm(OR)4n−2m], titanium-oxo-carboxo-alkoxide [TinO2n−x/2−y/2(OR)x(R′COO)y] and titanium-oxo-carboxo [TinOm(R′COO)p] clusters, as well as their syntheses, structures and NBB applications. In general, the construction of POT-based hybrid materials is built upon the possibility of modifying these preformed cages with polymerisable functional ligands (Fig. 2), by which the POT cages can be inter-connected through ligand-induced aggregation, crosslinking, and metal–organic-framework (MOF) formation (Fig. 2). These functional ligands, especially multidentate ones, can readily stabilize the cage against hydrolysis by substituting the more labile alkoxide groups. Such substitution is regioselective and can be carried out in a controlled manner,32,48 paving the way for the broad use of POTs in hybrid materials construction. The inter-cluster distance within these hybrids is ca. 1–5 nm and the electron density difference between the inorganic and organic constituents is large, making small-angle X-ray scattering (SAXS) a suitable method for their study.8 In addition to organic copolymers, hybrid materials can also be constructed by reacting L-POT cages with small molecules. For instance, using the alkyne–azide click reaction, the outward-pointing alkyne groups of [Ti6O4(OPr)8(OOC(CH2)2C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH)8] (3) can be employed as molecular ‘docks’ in reaction with azide groups to produce cluster-based inorganic–organic hybrids.49
CH)8] (3) can be employed as molecular ‘docks’ in reaction with azide groups to produce cluster-based inorganic–organic hybrids.49
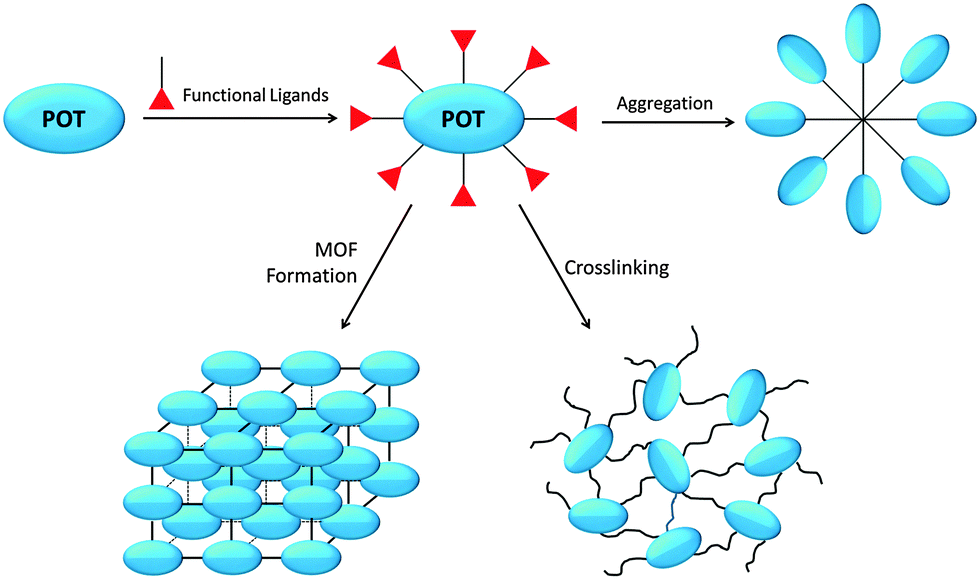 | ||
| Fig. 2 Schematic illustration of the construction of inorganic–organic hybrid materials from POT/L-POT cages. | ||
On a separate note, the concept of NBBs can be extended to the interaction of POT cages with macro-surfaces. Eslava et al.35 successfully modified the classic POT cage [Ti16O16(OEt)32] using the thiol–ether linker [HO(CH2)4SCH3] (Fig. 3). The resulting cage [Ti16O16(OEt)24{O(CH2)4SCH3}8] (4) was then covalently tethered onto an Au surface using five out of the eight thiol anchors in the cage (see Fig. 3). In contrast, the parent cage [Ti16O16(OEt)32] undergoes extensive decomposition on Au, revealing the importance of the soft thiol-modification for enhancing molecular stability and effective tethering onto the Au surface. Such surface tethering techniques could be of interest and have broad applications in sensors, heterocatalysis and other surface-based technologies.
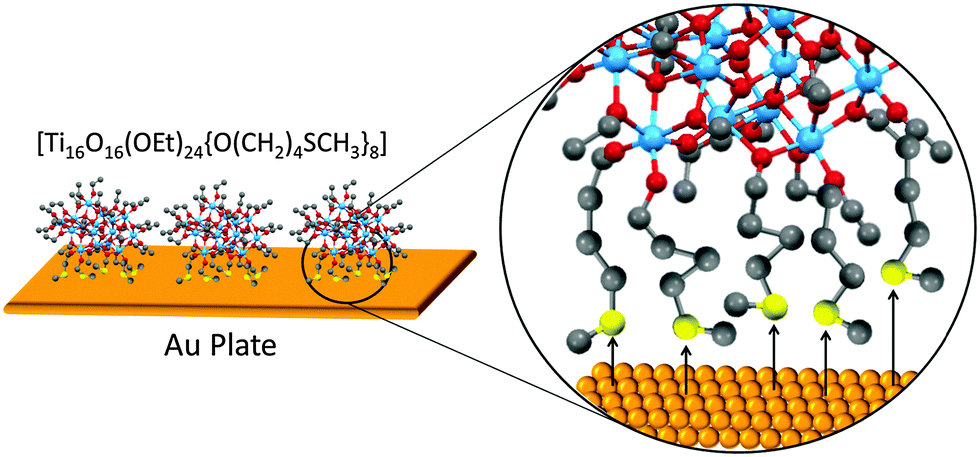 | ||
| Fig. 3 Schematic of the possible tethering mode of [Ti16O16(OEt)24{O(CH2)4SCH3}8] (4) onto an Au surface. Blue = Ti, red = O, grey = C, yellow = S, orange = Au. The Au–S interactions are indicated by black arrows. | ||
2.3 Photocatalysts
The general mechanism by which POT cages mediate photocatalysis involves the photo-induced electron excitation from the valence [predominantly O(p)] to the conduction band [mainly Ti(d)] of the TixOy core. The electrons and holes generated then migrate to the cage surface where redox reactions occur. However, band gaps (HOMO–LUMO) of unmodified POTs are usually in the range of 3.5–4.0 eV. This means that electronic excitation from the valence to conduction band requires photons of wavelength less than ca. 350 nm, ruling out >95% of the incident solar flux. While heterometallic doping has proved to be effective in narrowing the band gaps by introducing additional energy levels within the band gap of the POT core,7 functional ligand modification can be another important method of enhancing light absorption of POT systems.50 In general, functional ligands act as photo-sensitizers harvesting long wavelength photons, with the photoexcited electrons then being transferred to the POT core (Fig. 4).51 It is worth mentioning that the isolated ligand (L) itself does not necessarily absorb at the same energy as the L-POT. This is because, upon bonding with POTs, the ligand HOMO–LUMO can be mixed with the energy bands of the POT core, giving rise to a much narrower gap for the photoexcited electrons to overcome.31,52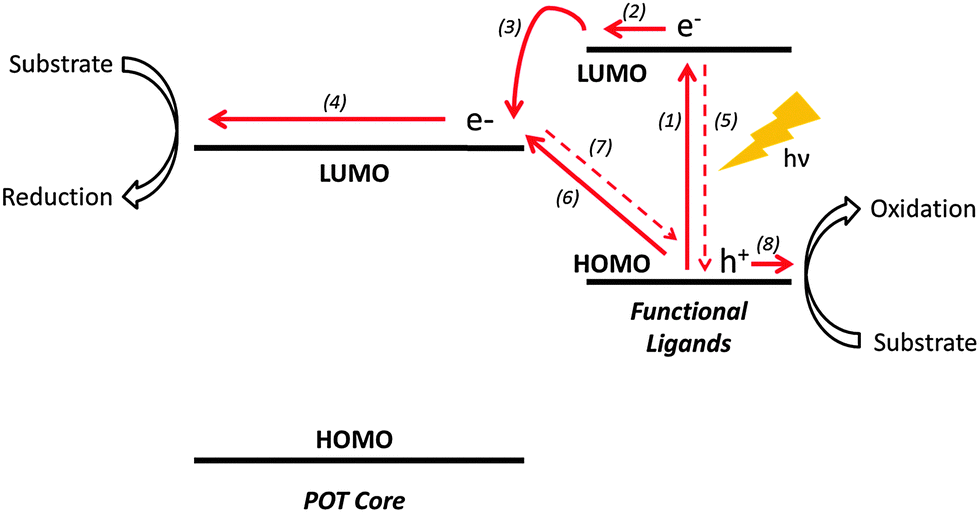 | ||
| Fig. 4 The mechanism of photocatalytic reactions mediated by L-POTs: (1) excitation of a functional ligand, (2) and (3) electron transfer from the excited functional ligand to the POT core, (4) electron transfer to the photocatalytic reaction substrate, (5) electron–hole recombination, (6) ligand-to-metal charge transfer, (7) electron–hole recombination, (8) hole transfer to the photocatalytic reaction substrate. | ||
One convenient way of studying the photo-responses of L-POTs is to deposit them onto transparent conducting substrates [e.g., fluorine-doped tin oxide (FTO), indium tin oxide (ITO)] and measuring the generated photocurrent upon light irradiation with or without applied voltages. However, it can be noted that the photocurrent response is not equivalent to photocatalytic activity, but merely an indication of it, as smaller HOMO–LUMO separation has to be balanced against the rate of hole–electron recombination. The optimum characteristics for high photocatalytic activity therefore include a reasonably small HOMO–LUMO gap and a slow recombination rate, so that visible light absorption is combined with the potential for hole–electron migration to the surface of the POT, where reactions with substrates can occur.
Using salicylic acid (SAL) and phthalic acid (o-BDC) as functional ligands, Hou et al.53 successfully obtained two loosely-packed L-POTs, [Ti13O10(o-BDC)4(SAL)4(OiPr)16] (5) (Fig. 5a) and [Ti13O10(o-BDC)4(SAL-Cl)4(OiPr)16] (6), and the corresponding photoelectrodes by spin-coating of these cages. The cage structures are retained on the electrode substrate (verified by the similarity of the IR spectra with the parent cages), and a clear photocurrent was observed, confirming the photo-response of these cage molecules (Fig. 5b). Despite the transparency of the utilized ligands themselves to visible light, both L-POTs show band gaps of ca. 2.5 eV that have been extended significantly into the visible region compared to the ca. 3.5 eV normally seen in unmodified POTs. Unfortunately, the magnitude of the photocurrent in both these cases is relatively small and the shape of the photocurrent plot is not an ideal square wave with cycled irradiation (Fig. 5b), possibly due to the non-conducting nature of the organic components.
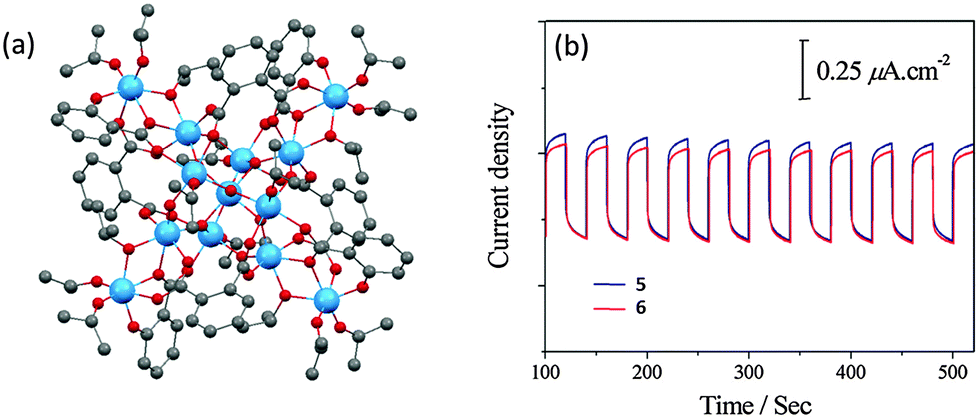 | ||
| Fig. 5 (a) Solid-state structure of [Ti13O10(o-BDC)4(SAL)4(OiPr)16] (5), blue = Ti, red = O, grey = C, hydrogen atoms are omitted for clarity. 6 is isostructural with 5, except that the –OH ortho hydrogen atoms are substituted by chlorine in four out of the six salicylate ligands; (b) photocurrent responses of the films of 5 and 6 on ITO upon cycling the irradiation. Panel (b) is reprinted with permission from ref. 53, ©2015 Royal Society of Chemistry. | ||
A similar photocurrent response has also been observed in 9-anthracenecarboxylic acid (9-AC) and catechol (cat) modified POTs.25 Owing to the catechol HOMO energy level mixing with that of POT core, crystalline blocks of [Ti6O4(OiPr)6(cat)4(9-AC)2] (7) are deep red in colour with a band gap of ca. 2.05 eV. Yet the photocurrent is far lower than that of [Ti6O6(OiPr)6(9-AC)6] (8) (Fig. 6), which only has 9-AC as functional ligands and exhibits a band gap of ca. 2.72 eV and is yellow in colour. This observation emphasises the point that efficient light absorption does not necessarily lead to a good photoelectrochemical response. Such counter-intuitive observations can be attributed to the retarded injection of photoexcited electrons from catechol to the cage core in 7. Thus, 8 shows a much larger photocurrent as it has more active 9-AC antennae for light harvesting. XPS spectra of a deposited film of 8 on ITO, both before and after light irradiation, revealed no TiIII signals, implying the instantaneous separation of the photogenerated electron–hole pairs. Another L-POT, [Ti6O3(OiPr)14(TTF)2]·0.5H2O (9) (TTF = dimethylthio-tetrathiafulvalene dicarboxylate), is also red in colour.24 However, rather than energy level penetration (as in cat-modified POTs), the red colour in 9 is attributed to π–π* excitation within the TTF ligand (absorption band at ca. 450 nm). More interestingly, the photocurrent density became much larger after exposing the clusters to air or oxygen, during which the red crystals gradually turned black (the colour of TTF radical cation, TTF+). Although the TTF ligands are retained after hydrolysis/oxidation (as confirmed by IR and solid-state CV), the OiPr groups are lost and cluster-to-cluster polymerization is observed. This may explain the photocurrent enhancement, as larger or extended TixOy cluster arrangements should be beneficial for charge transport.
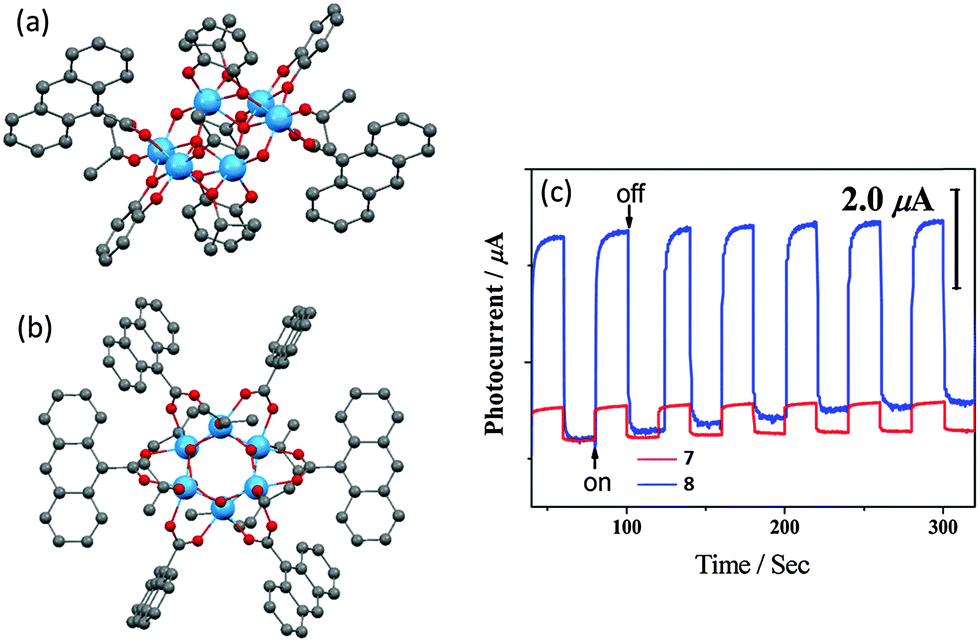 | ||
| Fig. 6 Solid-state structures of (a) [Ti6O4(OiPr)6(cat)4(9-AC)2] (7) and (b) [Ti6O6(OiPr)6(9-AC)6] (8), blue = Ti, red = O, grey = C, hydrogen atoms are omitted for clarity; (c) photocurrent responses of deposited films of 7 and 8 on ITO upon repeated cycles of irradiation. Panel (c) is reprinted with permission from ref. 25, ©2014 American Chemical Society. | ||
Metal complexes of 1,10-phenanthroline (Phen) represent one of the most important families of photosensitive dyes, giving rise to low band-gap solid-state materials or low HOMO–LUMO gap molecular complexes. However, its direct coordination to Ti centres is rare, and coordination of Phen to POTs has only been achieved on transition metal-doped M-POTs, in which Phen ligands all exclusively chelate the transition metal dopant ions.22,23,27 Dai and co-workers reported two Phen-modified M-POT cages, [Ti17O28(OiPr)16(CoPhen)2] (10) and [Ti17O28(OiPr)16(HOiPr)2(CdPhen)2] (11) (Fig. 7a).23 Coppens and co-workers27 enriched this family with an Fe analogue, [Ti17O28(OiPr)16(FePhen)2] (12). All these cages are isostructural except that the Cd centres in 11 are probably coordinated by two additional HOiPr ligands.23 The recently reported Phen-modified M-POT cage, [Ti18MnO30(OEt)20(MnPhen)3] (13) (Fig. 7b) exhibits completely different structural features to 10, 11 and 12, the three Phen ligands being coordinated to the three MnII centres to give a core structure with approximate C3v symmetry, with the fourth MnII ion being embedded within the core (tetra-coordinated by four oxo-O atoms).22
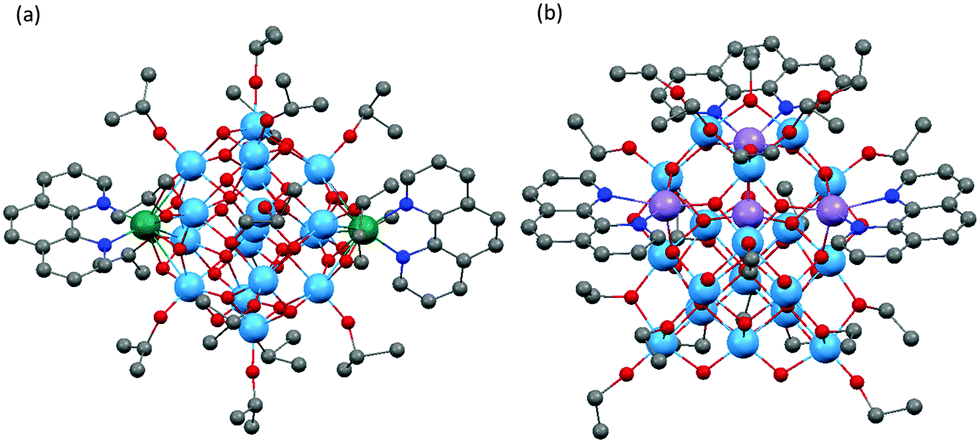 | ||
| Fig. 7 Solid-state structures of Phen-modified M-POTs: (a) [Ti17O28(OiPr)n(MIIPhen)2] (10, 11, 12) and (b) [Ti18MnO30(OEt)20(MnPhen)3] (13), blue = Ti, red = O, grey = C, green = Co, Fe or Cd, purple = Mn, dark blue = N. Hydrogen atoms are omitted for clarity. | ||
Due to the lack of direct bonding between Phen and Ti centres, it is difficult to investigate the physical effect of Phen coordination on POT cores themselves (i.e., in the absence of a transition metal ion). The photocurrent from these Phen-M-POT cages is transition-metal specific, the photocurrent being readily observed in the Co- and Fe-doped cages 10 and 12, but much weaker in the related Cd-doped counterpart 11, and completely absent from the Mn cage 13.22,23,27 The advantage of Co- and Fe-doped cages over the Cd counterpart probably simply stems from the greater redox activity of the transition metal ions. For instance, [Co(Phen)3]3+/2+ cations are a widely recognized photosensitizer, resulting from the reversible photoelectrochemical redox Co3+/Co2+ couple, so that an excited [Co(Phen)] fragment (in 10) can transfer electrons to the conduction band of the TixOy core upon irradiation.54 Similar behaviour can also be expected for [FePhen] fragments within cages like 12, but not for [CdPhen] since oxidization from Cd2+ to Cd3+ is of far too high energy. However, this does not explain the absence of a photocurrent for the [MnPhen]-containing cage 13, implying that photocurrent generation is a much more complicated process. Raman, IR and EDX data of the hydrolytic deposited films of 10 on an ITO substrate in the presence of ambient air show that this is [CoPhen]-decorated titania (i.e., that the structure of the complex is not retained).
Another important aspect of these L-POT deposited films is their self-assembled morphologies on FTO/ITO substrates. It is well-known that hollow-structured TiO2 with a high surface area, low density and high loading capacity can be used to improve the photon-to-energy conversion efficiency in solar cells and water splitting. However, due to the fast hydrolysis of titanium salts/alkoxides and the insolubility of TiO2, the fabrication of hollow nano-objects, such as hollow spheres, is a challenge without using a template or surfactant. Employing the L-POT cage 10 as a precursor, self-assembled microspheres or doughnut-like structures can be obtained simple by solution deposition (Fig. 8a and b).23 The possible mechanism of formation is illustrated schematically in Fig. 9. The deposited L-POT cage molecules quickly self-aggregate from solution upon evaporation of the solvent to form solvated spheres, the size of which is dependent on both the evaporation rate and solution concentration. As a result of atmospheric moisture, a shell of titania forms on the surface of the solvated spheres. With continuous hydrolysis and release of the inner solvent, the hollow spheres emerge. For larger spheres, part of the surface cannot be completed, which results in bowl- and doughnut-like structures.23 Other than 10, such microspheres have also been observed in the deposited films of 7 and 13,22,25 but not for other similar L-POT cages such as 5 and 8, which only show the formation of microplate morphology (Fig. 8c)25 or simply films without any special features (Fig. 8d).53 Although the detailed formation mechanism is not fully understood yet, there is no doubt that the morphology of deposited films is highly dependent on the L-POT precursor cage used, especially the presence of a functional ligand. This conclusion is further supported by the interesting behaviour of crystalline blocks of [Ti6O3(OiPr)14(TTF)2]·0.5H2O (9), which self-exfoliate upon exposure to air or oxygen, giving rise to stacks of ‘floorboard-like’ plates (Fig. 8e and f).24 The ready oxidation and exfoliation of the crystalline blocks can be attributed to catalysed reaction of 9 upon TTF oxidation and self-condensation of the POT core.
 | ||
| Fig. 8 SEM images of the deposited films from: (a) and (b) [Ti17O28(OiPr)16(CoPhen)2] (10); (c) [Ti6O6(OiPr)6(9-AC)6] (8) and (d) [Ti13O10(o-BDC)4(SAL)4(OiPr)16] (5); (e) the self-exfoliated crystal blocks of [Ti6O3(OiPr)14(TTF)2]·0.5H2O (9) and (f) side views showing the layered structure. Panels (a) and (b) are reprinted with permission from ref. 23, ©2013 Royal Society of Chemistry. Panels (c) and (d) are reprinted with permission from ref. 25, ©2014 American Chemical Society and ref. 53, ©2015 Royal Society of Chemistry. Panels (e) and (f) are reprinted with permission from ref. 24, ©2015 Royal Society of Chemistry. | ||
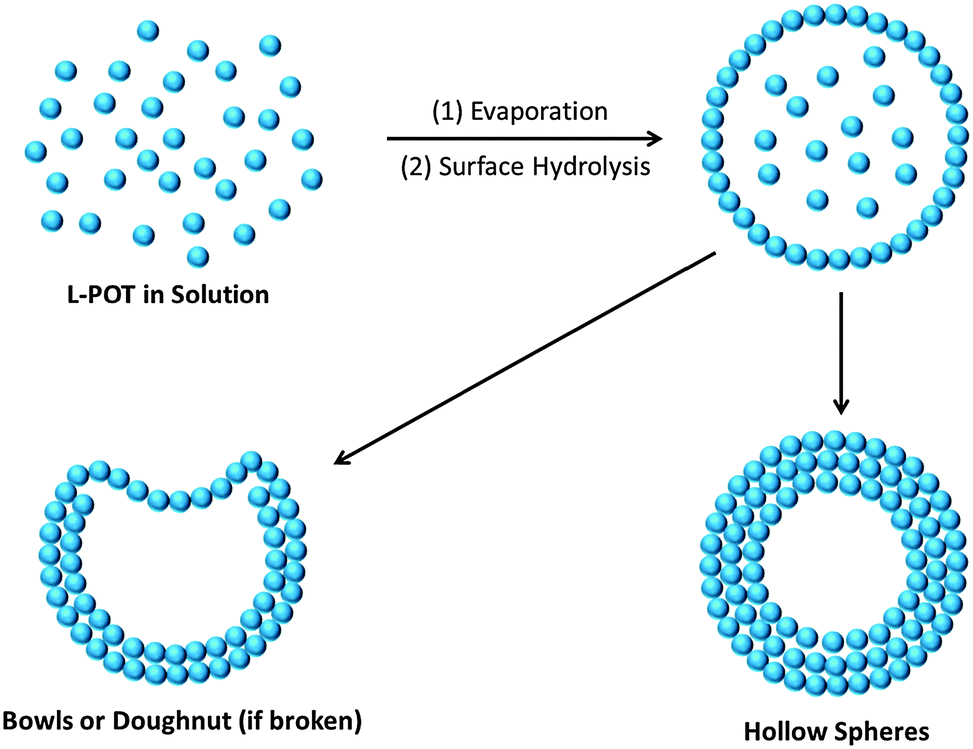 | ||
| Fig. 9 Schematic illustration of the formation mechanism of hollow microspheres, bowls or doughnuts from direct L-POT solution deposition. Each small sphere represents an L-POT cage. | ||
Moving from photocurrent response to the realms of photocatalytic activity itself, L-POT cages have been investigated in photocatalytic hydrogen evolution and organic pollutant photodegradation.28,55–58 Recently, Lin et al.55 synthesized three carboxylate-modified hexanuclear cages of the type [Ti6O4(OR)8(OOCR′)8] containing isostructural Ti6O4 cores. Under UV irradiation, suspensions consisting of these L-POTs, water and methanol (10%, as a sacrificial electron donor) produced gaseous hydrogen, with the highest rate of 326 μmol h−1 g−1 being found for the cage [Ti6O4(OEt)4(OiPr)4(OOCC6H5)8] (14) (Fig. 10a and c). This rate is comparable to (or even higher than) that found for nanostructured TiO2 counterparts. The identical XRD patterns before and after reaction confirm the stability of the cage structure of 14 in aqueous conditions, implying that the photocatalytic activity originates from the L-POT cages rather than any of potential hydrolysis products (such as titania).
 | ||
| Fig. 10 (a) Solid-state structures of (a) [Ti6O4(OEt)4(OiPr)4(OOCC6H5)8] (14) and (b) [Ti6O3(o-BDC)2(OiPr)14] (15), blue = Ti, red = O, grey = C, hydrogen atoms are omitted for clarity; (c) amount of H2 evolved vs. irradiation time for the cages of (14) (curve i), and the analogues [Ti6O4(OEt)8{OOC(CH3)3}8] (16) (curve ii) and [Ti6O4(OMe)8{OOC(CH3)3}8] (17) (curve iii); measurements were done in 100 mL of 10% methanol solution under UV irradiation with a 500 W mercury lamp; (d) photodegradation of MO vs. irradiation time for using 15 as a photocatalyst; measurement was done in 30 mL aqueous solution of methyl orange (0.1 mM) with 22 mg of suspended microcrystals of 15 and a trace amount of H2O2 (3%, 150 μL); (e) the proposed mechanism of MO photodegradation. Panel (c), is adapted with permission from ref. 55, ©2014 Elsevier. Panels (d) and (e) are adapted with permission from ref. 28, ©2012 American Chemical Society. | ||
The photodegradation of organic pollutants by L-POT cages is exemplified by a study in which the indicator methyl orange (MO) was degraded using white light irradiation in the presence of a UV filter (>420 nm) in an aqueous dispersion of microcrystals of [Ti6O3(o-BDC)2(OiPr)14] (15) (Fig. 10b).28 More than 90% decay of MO was observed after irradiation for 100 min (Fig. 10d). This photocatalytic effect is believed to be due to the formation of photo-induced O2˙− ions, and the proposed mechanism is shown in Fig. 10e.
2.4 Photochromism and photoluminescence
Photochromic behaviour has been observed for the cage [Ti6O4(o-BDC)2(o-BDC-iPr)2(OiPr)10] (18) (Fig. 11a), for which the colourless transparent crystals became purple-grey under UV-Vis irradiation (Fig. 11b and c).28 This colour change was attributed to the formation of photo-induced TiIII ions, which have been detected using ESR spectroscopy. No structural change for the crystal blocks was detected (by single crystal X-ray diffraction) after irradiation, indicating that the photochemical reaction only occurs on the surface. Although the UV-Vis spectrum of 18 after irradiation is not available, that of the analogous photochromic cage [Ti6O3(o-BDC)2(OiPr)14] (15) shows the formation of a new broad band at ca. 2.46 eV after irradiation (Fig. 11d), which can be assigned to the overlapped TiIII d–d and intervalence transitions. Back-oxidation of TiIII ions to TiIV could be achieved simply by exposing the coloured crystals to oxygen, which is associated with the reduction of molecular oxygen into superoxide ions O2˙− (confirmed by ESR). The detailed reaction route behind such photochromic behaviour is still elusive, but it seems that the overall cage structure (including a specific TixOy core and peripheral ligands) is crucial since no photochromism was observed for Ti(OiPr)4 under identical conditions and is also rare for other POT and L-POT cages. To our best knowledge, only one other example of photochromism has been shown in this area, the MOF [Ti8O8(OH)4(1,4-BDC)6] (19).59 Such photochromic behaviour should be of general interest, not only for the rational design of optical devices, but more importantly it underpins photocatalytic activities for POT cages and Ti-based MOF structures (e.g., oxidizing alcohols to aldehydes).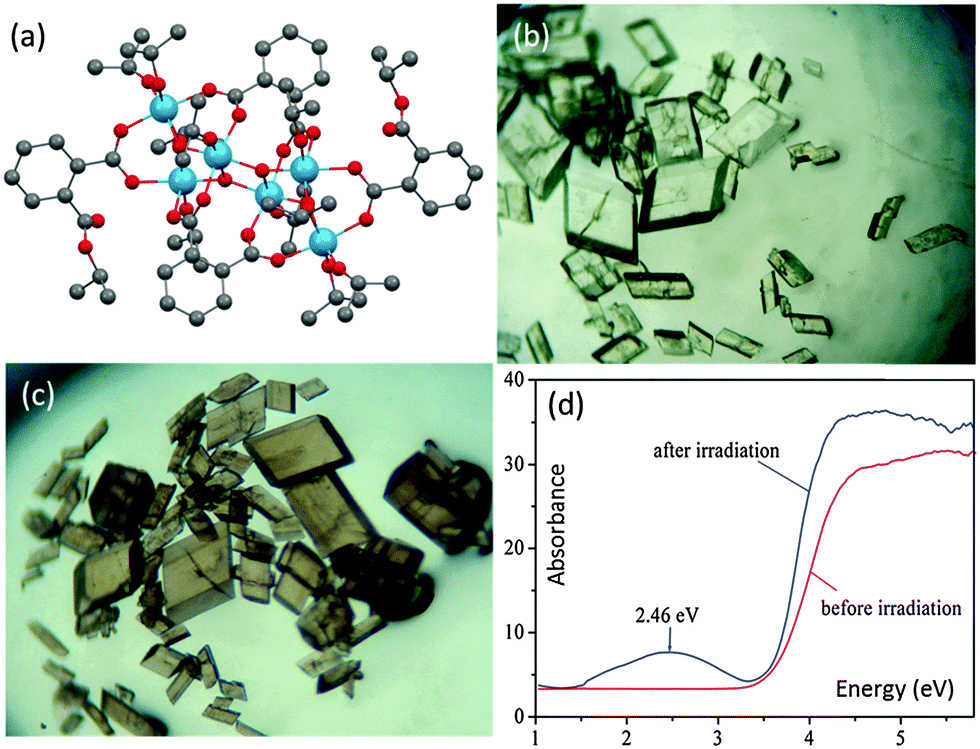 | ||
| Fig. 11 (a) Solid-state structure of [Ti6O4(o-BDC)2(o-BDC-iPr)2(OiPr)10] (18), blue = Ti, red = O, grey = C, hydrogen atoms are omitted for clarity; optical microscope images of crystalline blocks of 18 before (b) and after (c) irradiation; (d) UV-Vis spectra of an analogous photochromic L-POT [Ti6O3(o-BDC)2(OiPr)14] (15) before and after irradiation. Panels (b), (c) and (d) are reprinted with permission from ref. 28, ©2012 American Chemical Society. | ||
In contrast to photochromism, photoluminescence (PL) of L-POTs mainly arises from the peripheral functional ligands rather than the TixOy core. A clear PL signal has been observed for the 9-AC modified POT cage [Ti6O6(OiPr)6(9-AC)6] (8),25 consisting of a ‘stacked’ structure of two Ti3O3 ring units and six antenna 9-AC groups at its periphery (see Fig. 6b). The PL peak shifts from the characteristic emission of 9-AC at 465 nm to 457 nm (Fig. 12a), indicating charge-transfer from 9-AC to the cage core. Such charge-transfer is further confirmed by the lower PL quantum yield of 8 compared to the 9-AC ligand itself. As a result of solvent interactions with the frontier orbitals, the PL colour of 8 is closely related to the dipole moment of the solvents utilized, consequently CH2Cl2 (3.4 D), CH3CN (6.2 D) and H2O (10.2 D) give rise to the PL colours of green, blue and purple, respectively (Fig. 12c and d). Upon continuous irradiation, the PL intensity of 8 decreases and becomes completely extinct after ca. 60 min (Fig. 12b), but can be partially recovered very quickly by bubbling oxygen through the solution. Similar PL was also observed for [Ti6O4(OiPr)6(cat)4(9-AC)2] (7) (see Fig. 6a), which possesses a totally different cage core to 8, but with an identical bonding mode of the 9-AC ligands (bridging two adjacent Ti centres in the Ti6O4 cage core).25
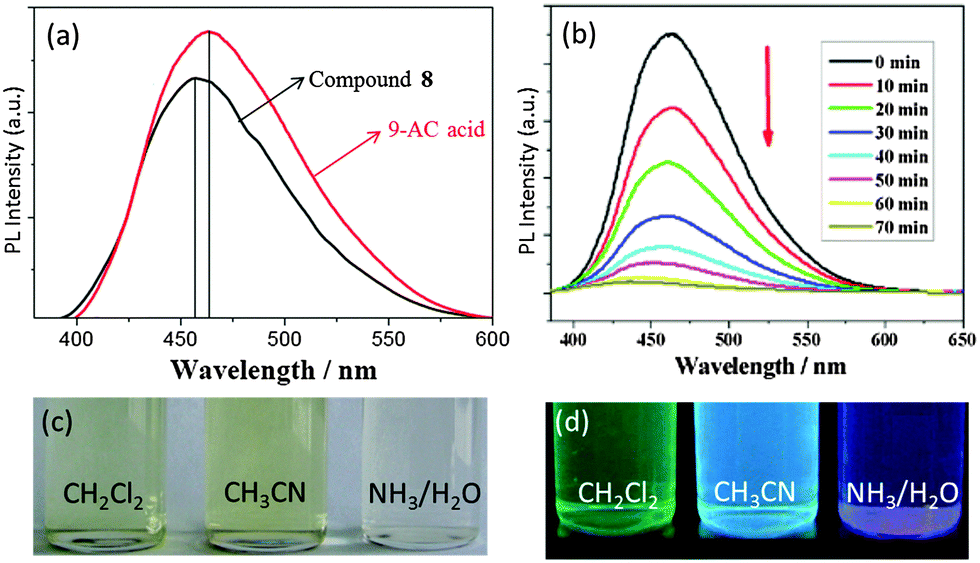 | ||
| Fig. 12 (a) PL emission spectra of cage 8 and 9-AC acid in CH2Cl2; (b) PL intensity of 8 in dichloromethane (1 × 10−4 mol L−1) upon continuous irradiation; digital photograph of a solution of 8 (c) and the PL colours (d) in CH2Cl2, CH3CN and diluted ammonia–water. Reprinted with permission from ref. 25, ©2014 American Chemical Society. | ||
In follow-up work, the 9-AC modified heterometallic POTs [Ti10Ln2O14(ClO4)2(OiPr)14(9-AC)2(CH3CN)2] [Ln = Nd (20) or Eu (21)] (Fig. 13a) were isolated.26 These two isostructural cages demonstrate similar PL spectra, with band maxima at ca. 460 nm (Fig. 13c), but the intensity of 20 is about 0.6 times higher than that from 21 under identical conditions. Another two analogous cages, [Ti10Ln2O14(ClO4)2(OiPr)14(bza)2(HOiPr)2] (Ln = Nd (22) or Eu (23), bza = benzoate) (Fig. 13b) with the 9-AC ligands replaced by PL-inactive benzoate, were also prepared in order to explore the detailed PL mechanism. Compound 23 exhibits a typical EuIII-centred red emission (Fig. 13d). Such emission is completely absent from 21, which only shows a single band of ligand-centred emission at 460 nm (Fig. 13c and d). Even coupled with a 540 nm filter to minimize the influences of the strong ligand-centred PL signal, the Eu-centre emission was still absent (Fig. 13e). This absence can be explained by the mismatch of energy levels, as the triplet energy level of 9-AC is lower than the Eu-centred excited levels. A diagram of the proposed energy-transfer process for 21 is depicted in Fig. 13f, which explains not only the absence of the EuIII-centred PL signal but also the enhanced ligand-centred PL intensities of 21 over 20. When irradiated at 320 nm, the EuIII-doped POT core of 21 is excited. The electron energy then transfers from the cage conduction band to the S1 state of 9-AC, which enhances the ligand-centred emission. Moreover, the long lifetime of the 5D excited state of EuIII also plays an important role in this energy-transfer process. This provides a possible reason why such an energy-transfer process does not occur in the Nd cage 20, as the lifetime of NdIII excited state is only 0.2–0.5 ms, significantly shorter than that of EuIII (1–11 ms).
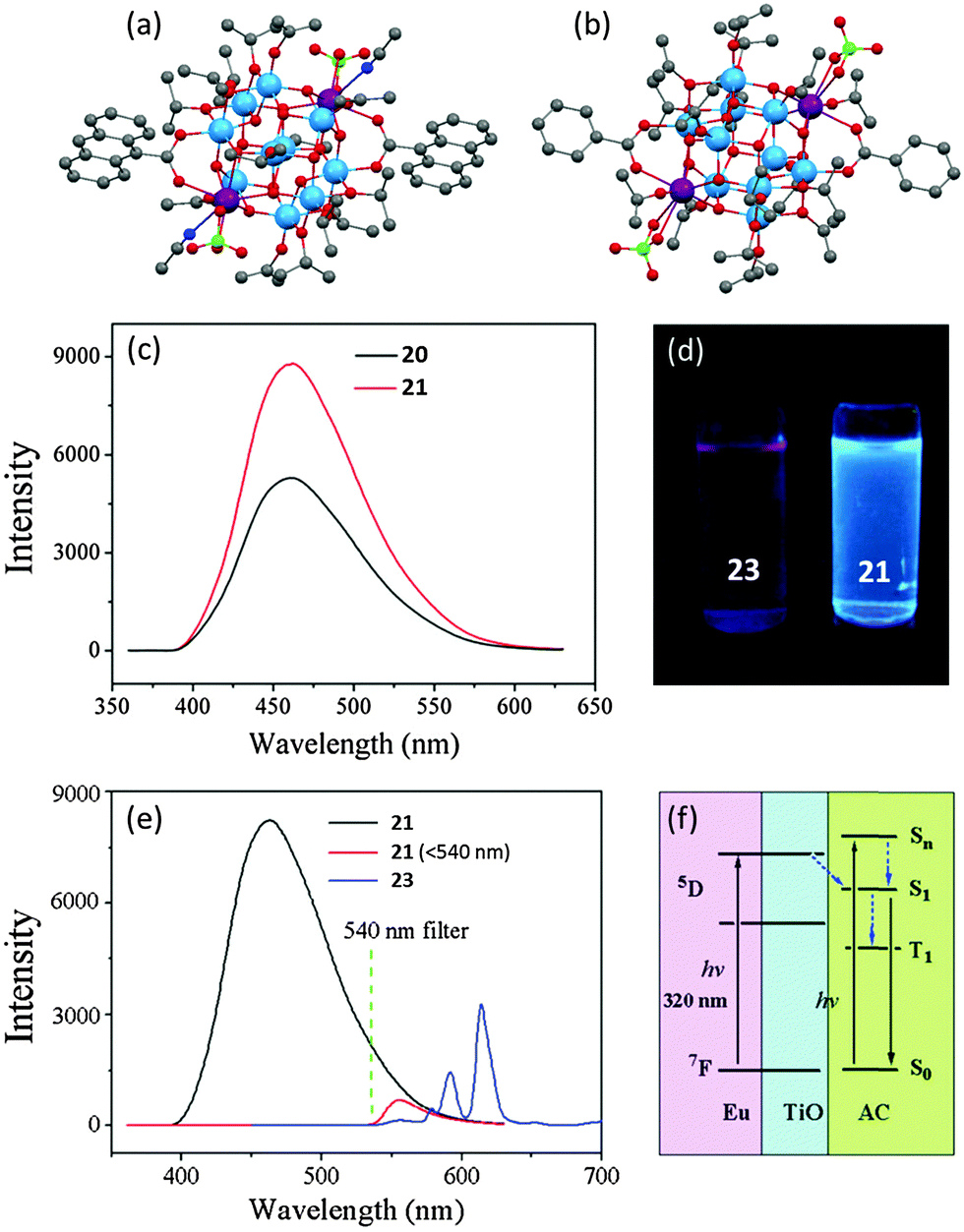 | ||
Fig. 13 Solid-state structures of compound (a) 20 and 21, (b) 22 and 23, blue = Ti, purple = Nd or Eu, red = O, grey = C, green = Cl, dark blue = N; (c) PL spectra of compound 20 and 21 in a mixed solvent of methanol and dichloromethane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 v/v) with concentration of 0.1 mM; (d) digital photograph of PL colours of 23 (left) and 21 (right); (e) PL spectra of 21 and 23 with and without a 540 nm optical filter; (f) proposed energy transfer mechanism in 21. Panels (c), (d), (e) and (f) are reprinted with permission from ref. 26, ©2015 Royal Society of Chemistry. :5 v/v) with concentration of 0.1 mM; (d) digital photograph of PL colours of 23 (left) and 21 (right); (e) PL spectra of 21 and 23 with and without a 540 nm optical filter; (f) proposed energy transfer mechanism in 21. Panels (c), (d), (e) and (f) are reprinted with permission from ref. 26, ©2015 Royal Society of Chemistry. | ||
2.5 Asymmetric catalysts
Catalysts based on POTs, especially those coordinated with 1,1′-bi-2-naphthol (BINOL) and their derivatives, have been applied to a number of enantioselective carbon–carbon bond-forming reactions, such as oxidation of thilanisole, cyanosilylation of aldehydes, Diels–Alder cycloaddition, carbonyl–ene reactions, nitro-aldol reactions, asymmetric allylation of aldehydes/ketones and asymmetric sulphide oxidation.60,61 The Ti–BINOL catalysed reactions can undergo a ligand-accelerated catalysis process that involves in situ selection of the active species from many thermodynamically accessible complexes present, even though the catalytically-active species may be present only in a small amount.62 This makes it difficult to identify and isolate the actual catalytically-active species. As a result, so far structural information on the real catalysts in this area is largely lacking. For example, the mononuclear complex [Ti(BINOLato)(OiPr)2] (24) is a pre-catalyst for many reactions, but the complex itself is not the active species.63The literature involving structurally-characterised Ti–BINOL catalysts is relatively sparse60,64–69 compared to the large amount of reports investigating their catalytic performance. Among the catalysts with known structures, the tetranuclear cages [Ti4(μ3-OH)4(μ-BINOLato)6] (R-BINOLato for 25, S-BINOLato for 26) are of particular interest (Fig. 14a), as they are extremely stable, even in strongly acidic or basic conditions under reflux. All six BINOLato ligands within 25 and 26 have the same stereochemistry, either R or S, bonding with the distorted cubic cage core via Ti–O bonds. Mahrwald et al.66 have found 26 to be effective in the catalysis of aldol addition with high regioselectivity at the more steric α-encumbered side of unsymmetrical ketones (Scheme 3). Catalyst loadings as low as 0.2 mol% were enough to afford complete conversion very smoothly without the formation of a significant amount of byproducts. Moreover, the tetranuclear cages [Ti4(μ3-OH)4(7,7′-di-R-(R)-BINOLato)6] (R = H for 27, Br for 28, BnO for 29, Ph for 30, PhCC for 31) have been found to catalyse [2+3] cycloaddition reactions (Scheme 4).69 The introduction of the sterically-bulky 7,7′-Ph-substituent leads to an increase in enantioselectivity for the exo-product (Scheme 4). In the exo-transition state, tert-butyl vinyl ether approaches from the Re-face of the nitrone preferentially, affording the 3R,5S product because access to the Si-face (leading to the 3S,5R product) is sterically hindered by the bulky 7,7′-substituents (Fig. 14c).69
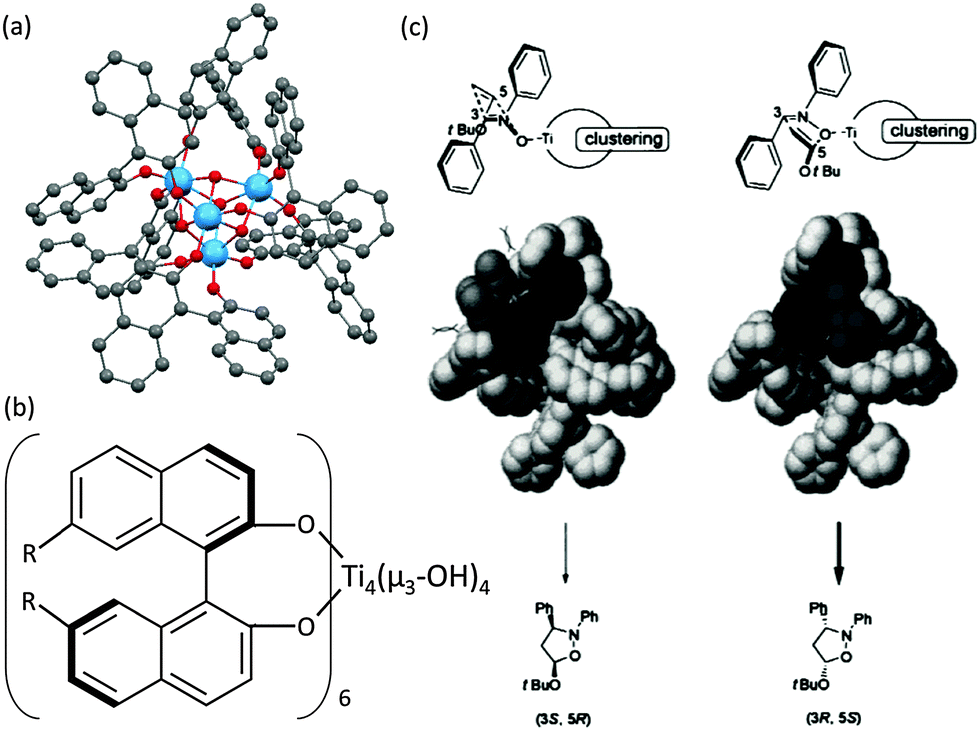 | ||
| Fig. 14 (a) Molecular structure of the tetranuclear [Ti4(μ3-OH)4(R-BINOLato)6] (25). The S-BINOL enantiomer structure has a similar configuration (not shown); (b) molecular representation of 7,7′-substituted [Ti4(μ3-OH)4(7,7′-di-R-BINOLato)6] cage; (c) illustration of the steric reason for the favourable production of (3R,5S) over (3S,5R) products. Panel (c) is adapted with permission from ref. 69, ©2001 Wiley. | ||
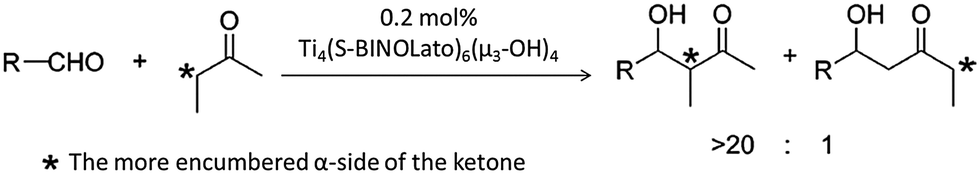 | ||
| Scheme 3 Direct aldol addition catalysed by [Ti4(μ3-OH)4(S-BINOLato)6] (26) with high regioselectivity at the more steric α-encumbered side of unsymmetrical ketones. Adapted with permission from ref. 66, ©2006 American Chemical Society. | ||
 | ||
| Scheme 4 The [2+3] cycloaddition reaction catalysed by [Ti4(μ3-OH)4(7,7′-di-R-(R)-BINOLato)6]. The table shows the catalytic capability of different 7,7′-substituted compounds. Adapted with permission from ref. 69, ©2001 Wiley. | ||
2.6 Gas adsorption
Crystalline blocks of metal-oxo cages can be described as molecular solids, owing to the absence of intermolecular covalent or coordination bonds between them that would form extended network structures, as is the case for MOFs. Such molecular solids possess clear advantages as potential gas adsorbents over many other physisorption materials, including: (i) they do not lose sorption properties upon amorphization (unlike most MOFs) and therefore can be utilized under a wide range of conditions, (ii) being inherently discrete (molecular) in nature, they are soluble and therefore can readily be modified or processed into other forms using solid or solution techniques, and (iii) their facile synthesis and vast structural diversity is nearly comparable to those of MOFs.70POT cages, especially the structurally-diverse carboxylate-modified ones, are good candidates as gas adsorbents.71,72 In general, densely-packed crystals are stable against thermal treatment and desolvation, unlike less densely packed lattices which lose their crystallinity after similar treatment.72 For example, solid [Ti6O6(abz)6(OiPr)6] (abz = aminobenzoate) (32) (Fig. 15a), with only 10% of solvent-accessible void volume in the lattice, maintains its crystallinity after heating to 200 °C, whereas under identical conditions the compound [Ti8O10(abz)12] (33) (Fig. 15b) (with 33% solvent-accessible lattice void) loses its crystallinity. Although neither 32 nor 33 demonstrate any N2 or CH4 adsorption properties, a significant uptake of CO2 at 195 K was observed for both compounds (Fig. 15c and d), possibly due to the small kinetic diameter of CO2 (ca. 3.3 Å) and polar nature of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond which facilitates interaction with the polar regions of the solids.72 Interestingly, after complete amorphitization of the crystallinity of 32, its CO2 adsorption capability is still retained (inset to Fig. 15c), which shows that the selective and reversible adsorption of gaseous CO2 by molecular carboxylate-POTs even without intramolecular pores can occur exclusively through intermolecular voids and that it is irrelevant whether the sample is crystalline or not.
O bond which facilitates interaction with the polar regions of the solids.72 Interestingly, after complete amorphitization of the crystallinity of 32, its CO2 adsorption capability is still retained (inset to Fig. 15c), which shows that the selective and reversible adsorption of gaseous CO2 by molecular carboxylate-POTs even without intramolecular pores can occur exclusively through intermolecular voids and that it is irrelevant whether the sample is crystalline or not.
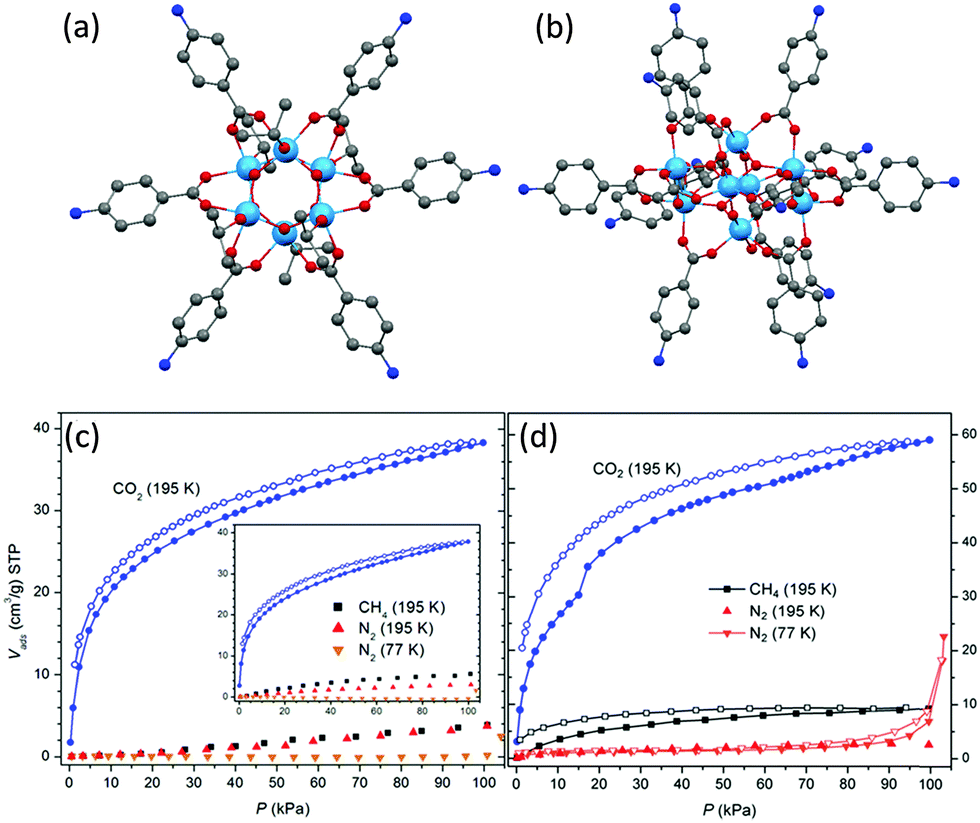 | ||
| Fig. 15 Solid-state structures of (a) [Ti6O6(abz)6(OiPr)6] (32) and (b) [Ti8O10(abz)12] (33) (abz = aminobenzoate), blue = Ti, red = O, blue = N, grey = C, hydrogen atoms are omitted for clarity. Gas-sorption isotherms measured for compound (c) 32 and (d) 33; inset of (c) is the isotherms of 32 with complete crystalline disruption; filled and open symbols denote adsorption and desorption, respectively. Panels (c) and (d) are reprinted with permission from ref. 72, ©2013 American Chemical Society. | ||
The specific packing of the molecules in the crystalline solids does have an apparent impact on CO2 adsorption, although this has little effect in the case of 32 and 33 (Fig. 15c and d). In general, the adsorption in loosely-packed compounds, such as is seen in the lattice of [Ti6O6(4-tbbz)6(OiPr)6] (4-tbbz = 4-tert-butyl-benzoate) (34, Fig. 16a), can occur in the intermolecular voids without significantly disturbing the molecular arrangement. Here a typical absorption type I curve is observed without hysteresis between the adsorption and desorption (Fig. 16e, blue curve for 34). While in a densely-packed lattice as is the case for [Ti6O6(4-tbbz)10(OiPr)2] (35, Fig. 16b), the adsorption of gas molecules can only occur with the expansion of the lattice parameters. Such structural expansion is believed to be responsible for the distinct step in the adsorption branch of CO2 for 35 (Fig. 16e).71 Further studies have revealed that the substituents on the carboxylate ligands also play an important role in the adsorption of gaseous CO2.71 By using relatively linear carboxylate ligands (e.g., 4-tbbz, Fig. 16c), the intermolecular voids between adjacent ligands can be preserved or even enlarged. As a result, cages bearing such ligands (such as 34 and 35) can adsorb almost the same number of CO2 molecules (ca. 5.5 per cage) despite the different molecular geometries and crystal packings present (Fig. 16e). The cages [Ti6O6(2-bpyc)6(OiPr)6] (36) and [Ti6O6(2-bpyc)10(OiPr)2] (37), in which the linear 4-tbbz ligands have been replaced by non-linear and bulky biphenyl-2-carboxylate (2-bpyc, Fig. 16d), are isostructural with 34 and 35. However, they exhibit a far lower CO2 adsorption capacity of approximately one CO2 molecule per cage (Fig. 16c, red curves). This can be ascribed to the fact that the phenyl group at the 2-position of the carboxylate ligand occupies the intermolecular space between the clusters in the lattice, resulting in the blocking of the voids where the adsorption of CO2 occurs.
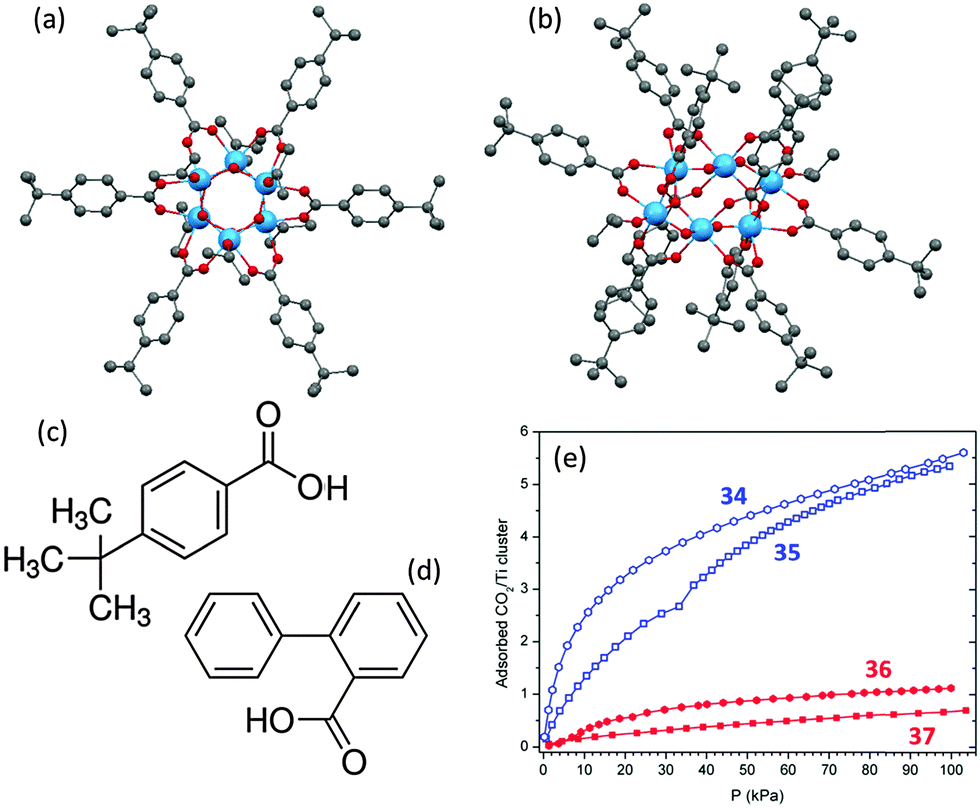 | ||
| Fig. 16 Solid-state structures of (a) [Ti6O6(4-tbbz)6(OiPr)6] (34) and (b) [Ti6O6(4-tbbz)10(OiPr)2] (35) (4-tbbz = 4-tert-butyl-benzoate), blue = Ti, red = O, grey = C, hydrogen atoms are omitted for clarity. Molecular structure of the functionalized ligands (c) 4-tert-butyl-benzoic acid (4-tbbz) and (d) biphenyl-2-carboxylic acid (2-bpyc); (f) CO2 adsorption isotherms for 34, 35, 36 and 37 with Y axis being the number of TiO2 molecules adsorbed per cluster. Panel (e) is adapted with permission from ref. 71, ©2014 American Chemical Society. | ||
An interesting two-step adsorption profile of CO2 has been observed for titanium cages that possess both intermolecular voids as well as intramolecular pores.72 For example, with the CO2 pressure below a critical value (P/P0 ∼ 0.6), perfectly reversible type I adsorption is observed for the cyclic cage [Ti8O10(benzoate)12] (38) (Fig. 17), because of the presence of permanent intramolecular pores of ca. 4 Å in diameter. The almost saturated adsorption then shows a sudden increase with large hysteresis at higher pressures (Fig. 17b). This second step is due to adsorption into the lattice voids that interconnect adjacent ring molecules. Also of note is the non-selective sorption of N2 and CH4 at 195 K for 38, again due to the presence of the permanent intramolecular pore in the molecule (Fig. 17b). Relatively higher absorption of CH4 compared to N2 at the same temperature is a generally observed phenomenon that originates from the greater polarizability of CH4 (2.59 Å3 for CH4vs. 1.74 Å3 for N2), which leads to stronger interactions with molecular solids.
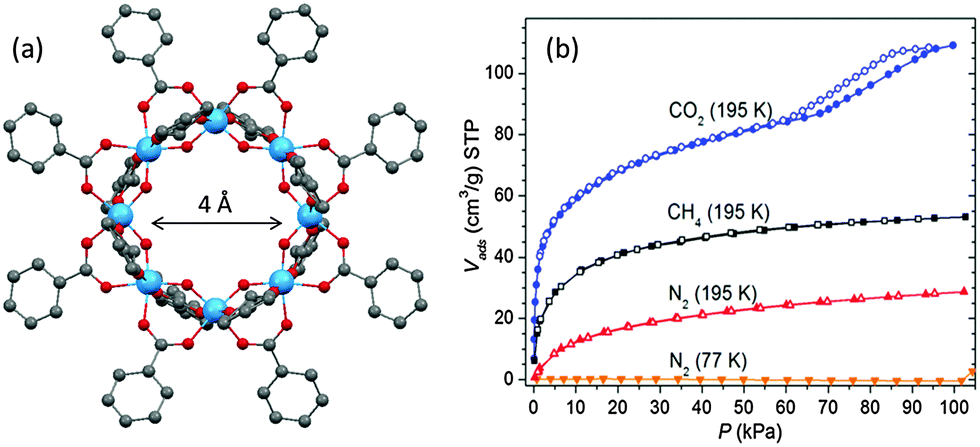 | ||
| Fig. 17 (a) Solid-state structure of [Ti8O10(benzoate)12] (38) with permanent inner pore of ca. 4 Å in size, blue = Ti, red = O, grey = C, hydrogen atoms are omitted for clarity; (b) gas-sorption isotherms of 38; filled and open symbols denote adsorption and desorption, respectively. Panel (b) is reprinted with permission from ref. 72, ©2013 American Chemical Society. | ||
3. Summary and perspective
This article has highlighted recent advances of functional ligand-modified polyoxotitanate (L-POT) cages, focusing specifically on novel properties and potential applications in a broad range of fields. Compared to the POTs and M-POTs that have mainly been used as single-source precursors for TiO2/TiO2(M) deposition, the introduction of functional ligands introduces more diverse properties either solely from the functional ligands themselves or synergistically as a result of the combination of the properties of the functional ligand (L) with the POT core. The sheer range of potential applications of L-POTs, which spans the areas of supramolecular chemistry, photocatalysis, photochromism photoluminescence, asymmetric catalysis and gas adsorption, makes them an almost unique family of compounds with exceptional future promise in a number of technological fields.Acknowledgements
We thank the AGSO (scholarship for N. L.), EPSRC (studentship for P. D. M.), and the ERC (Advanced Investigator Grant, D. S. W.) for financial support.Notes and references
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891 CrossRef CAS PubMed.
- G. Pfaff and P. Reynders, Chem. Rev., 1999, 99, 1963 CrossRef CAS PubMed.
- J. Bai and B. Zhou, Chem. Rev., 2014, 114, 10131 CrossRef CAS PubMed.
- J. Nisar, Z. Topalian, A. D. Sarkar, L. Österlund and R. Ahuja, ACS Appl. Mater. Interfaces, 2013, 5, 8516 CAS.
- U. I. Gaya and A. H. Abdulah, J. Photochem. Photobiol., C, 2008, 9, 1 CrossRef CAS.
- M. Pelaez, N. T. Nolan, S. C. Pillai, M. K. Seery, P. Falaras, A. G. Kontos, P. S. M. Dunlop, J. W. J. Hamilton, J. A. Byrne, K. O'Shea, M. H. Entezari and D. D. Dionysiou, Appl. Catal., B, 2012, 125, 331 CrossRef CAS.
- P. D. Matthews, T. C. King and D. S. Wright, Chem. Commun., 2014, 50, 12815 RSC.
- L. Rozes and C. Sanchez, Chem. Soc. Rev., 2011, 40, 1006 RSC.
- P. Coppens, Y. Chen and E. Trzop, Chem. Rev., 2014, 114, 9645 CrossRef CAS PubMed.
- J. B. Benedict, R. Freindorf, E. Trzop, J. Cogswell and P. Coppens, J. Am. Chem. Soc., 2010, 132, 13669 CrossRef CAS PubMed.
- L. Wu, J. C. Yu, L. Zhang, X. Wang and W. Ho, J. Solid State Chem., 2004, 177, 2584 CrossRef CAS.
- S. Eslava, B. P. R. Goodwill, M. McPartlin and D. S. Wright, Inorg. Chem., 2011, 50, 5655 CrossRef CAS PubMed.
- Y. Lv, J. Cheng, P. D. Matthews, J. P. Holgado, J. Willkomm, M. Leskes, A. Steiner, D. Fenske, T. C. King, P. T. Wood, L. Gan, R. M. Lambert and D. S. Wright, Dalton Trans., 2014, 43, 8679 RSC.
- S. Eslava, M. McPartlin, R. I. Thomson, J. M. Rawson and D. S. Wright, Inorg. Chem., 2010, 49, 11532 CrossRef CAS PubMed.
- Y. Lv, J. Cheng, A. Steiner, L. Gan and D. S. Wright, Angew. Chem., Int. Ed., 2014, 53, 1934 CrossRef CAS PubMed.
- S. Eslava, F. Hengesbach, M. McPartlin and D. S. Wright, Chem. Commun., 2010, 46, 4701 RSC.
- Y.-H. Lai, C.-Y. Lin, Y. Lv, T. C. King, A. Steiner, N. M. Muresan, L. Gan, D. S. Wright and E. Reisner, Chem. Commun., 2013, 49, 4331 RSC.
- Y. Lv, J. Willkomm, A. Steiner, L. Gan, E. Reisner and D. S. Wright, Chem. Sci., 2012, 3, 2470 RSC.
- Y.-H. Lai, T. C. King, D. S. Wright and E. Reisner, Chem. – Eur. J., 2013, 19, 12943 CrossRef CAS PubMed.
- Y. Lv, J. Willkomm, M. Leskes, A. Steiner, T. C. King, L. Gan, E. Reisner, P. T. Wood and D. S. Wright, Chem. – Eur. J., 2012, 18, 11867 CrossRef CAS PubMed.
- Y. Lv, M. Yao, J. P. Holgado, T. Roth, A. Steiner, L. Gan, R. M. Lambert and D. S. Wright, RSC Adv., 2013, 3, 13659 RSC.
- N. Li, P. D. Matthews, J. J. Leung, T. C. King, P. T. Wood, H.-K. Luo and D. S. Wright, Dalton Trans., 2015, 44, 19090 RSC.
- Y.-Y. Wu, P. Wang, Y.-H. Wang, J.-B. Jiang, G.-Q. Bian, Q.-Y. Zhu and J. Dai, J. Mater. Chem. A, 2013, 1, 9862 CAS . The original report of 11 gives its formula as [Ti17O28(OiPr)18(CdPhen)2], which is clearly in error as the charge of this complex would be −2.
- J.-X. Yin, P. Huo, S. Wang, J. Wu, Q.-Y. Zhu and J. Dai, J. Mater. Chem. C, 2015, 3, 409 RSC.
- Y.-Y. Wu, X.-W. Lu, M. Qi, H.-C. Su, X.-W. Zhao, Q.-Y. Zhu and J. Dai, Inorg. Chem., 2014, 53, 7233 CrossRef CAS PubMed.
- S. Wang, H.-C. Su, L. Su, Y.-W. Zhao, L.-W. Qian, Q.-Y. Zhu and J. Dai, Dalton Trans., 2015, 44, 1882 RSC.
- K. N. Jarzembska, Y. Chen, J. N. Nasca, E. Trzop, D. F. Watson and P. Coppens, Phys. Chem. Chem. Phys., 2014, 16, 15792 RSC.
- Y.-Y. Wu, W. Luo, Y.-H. Wang, Y.-Y. Pu, X. Zhang, L.-S. You, Q.-Y. Zhu and J. Dai, Inorg. Chem., 2012, 51, 8982 CrossRef CAS PubMed.
- R. C. Snoeberger III, K. J. Young, J. Tang, L. J. Allen, R. H. Crabtree, G. W. Brudvig, P. Coppens, V. S. Batista and J. B. Benedict, J. Am. Chem. Soc., 2012, 134, 8911 CrossRef PubMed.
- J. D. Sokolow, E. Trzop, Y. Chen, J. Tang, L. J. Allen, R. H. Crabtree, J. B. Benedict and P. Coppens, J. Am. Chem. Soc., 2012, 134, 11695 CrossRef CAS PubMed.
- J. B. Benedict and P. Coppens, J. Am. Chem. Soc., 2010, 132, 2938 CrossRef CAS PubMed.
- G. Fornasieri, L. Rozes, S. L. Calvé, B. Alonso, D. Massiot, M. N. Rager, M. Evain, K. Boubekeur and C. Sanchez, J. Am. Chem. Soc., 2005, 127, 4869 CrossRef CAS PubMed.
- C. Chaumont, E. Huen, C. Huguenard, P. Mobian and M. Henry, Polyhedron, 2013, 57, 70 CrossRef CAS.
- I. Mijatovic, G. Kickelbick, M. Puchberger and U. Schubert, New J. Chem., 2003, 27, 3 RSC.
- S. Eslava, A. C. Papageorgiou, S. K. Beaumont, G. Kyriakou, D. S. Wright and R. M. Lambert, Chem. Mater., 2010, 22, 5174 CrossRef CAS.
- A. Rammal, F. Brisach and M. Henry, C. R. Chimie, 2002, 5, 59 CrossRef CAS.
- T. J. Boyle, L. J. Tribby, T. M. Alam, S. D. Bunge and G. P. Holland, Polyhedron, 2005, 24, 1143 CrossRef CAS.
- T. J. Boyle, R. P. Tyner, T. M. Alam, B. L. Scott, J. W. Ziller and B. G. Potter Jr., J. Am. Chem. Soc., 1999, 121, 12104 CrossRef CAS.
- I. Karatchevtseva, D. J. Cassidy, Z. Zhang, G. Triani, K. S. Finnie, S. L. Cram and C. J. Barbé, J. Am. Ceram. Soc., 2008, 91, 2015 CrossRef CAS.
- A. Mehner, T. Rüffer, H. Lang, A. Pohlers, W. Hoyer and S. Spange, Adv. Mater., 2008, 20, 4113 CrossRef CAS.
- G. L. Puma, A. Bono, D. Krishnaiah and J. G. Collin, J. Hazard. Mater., 2008, 157, 209 CrossRef PubMed.
- K. Black, A. C. Jones, J. Bacsa, P. R. Chalker, P. A. Marshall, H. O. Davis, P. N. Heys, P. O'Brien, M. Afzaal, J. Raftery and G. W. Critchlow, Chem. Vap. Deposition, 2010, 16, 93 CrossRef CAS.
- N. Hollingsworth, M. Kanna, G. Kociok-Köhn, K. C. Molloy and S. Wongnawa, Dalton Trans., 2008, 631 RSC.
- A. Chemseddine and T. Moritz, Eur. J. Inorg. Chem., 1999, 235 CrossRef CAS.
- G. Férey, Chem. Soc. Rev., 2008, 37, 191 RSC.
- N. Miele-Pajot, L. G. Hubert-Pfalzgraf, R. Papiernik, J. Vaissermann and R. Collier, J. Mater. Chem., 1999, 9, 3027 RSC.
- N. Steunou, S. Förster, P. Florian, C. Sanchez and M. Antonietti, J. Mater. Chem., 2002, 12, 3426 RSC.
- T. Frot, S. Cochet, G. Laurent, C. Sassoye, M. Popall, C. Sanchez and L. Rozes, Eur. J. Inorg. Chem., 2010, 5650 CrossRef CAS.
- P. Heinz, M. Puchberger, M. Bendova, S. O. Baumann and U. Schubert, Dalton Trans., 2010, 39, 7640 RSC.
- J.-X. Liu, M.-Y. Gao, W.-H. Fang, L. Zhang and J. Zhang, Angew. Chem., Int. Ed., 2016, 55, 5160 CrossRef CAS PubMed.
- G. Zhang, G. Kim and W. Choi, Energy Environ. Sci., 2014, 7, 954 CAS.
- C. Chaumont, P. Mobian and M. Henry, Dalton Trans., 2014, 43, 3416 RSC.
- J.-L. Hou, W. Lou, Y.-Y. Wu, H.-C. Su, G.-L. Zhang, Q.-Y. Zhu and J. Dai, Dalton Trans., 2015, 44, 19829 RSC.
- S. M. Feldt, E. A. Gibson, E. Gabrielsson, L. Sun, G. Boschloo and A. Hagfeldt, J. Am. Chem. Soc., 2010, 132, 16714 CrossRef CAS PubMed.
- Y. Lin, Y.-F. Zhu, Z.-H. Chen, F.-H. Liu, L. Zhao and Z.-M. Su, Inorg. Chem. Commun., 2014, 40, 22 CrossRef CAS.
- L. Ni, D. Liang, Y. Cai, G. Diao and Z. Zhou, Dalton Trans., 2016, 45, 7581 RSC.
- J.-X. Liu, X.-C. Zeng, L. Zhang and J. Zhang, Dalton Trans., 2016, 45, 4501 RSC.
- W. Luo and G. Ge, J. Cluster Sci., 2016, 27, 635 CrossRef CAS.
- M. Dan-Hardi, C. Serre, T. Frot, L. Rozes, G. Maurin, C. Sanchez and G. Férey, J. Am. Chem. Soc., 2009, 131, 10857 CrossRef CAS PubMed.
- K. Mikami, Y. Matsumoto and L. Xu, Inorg. Chim. Acta, 2006, 359, 4159 CrossRef CAS.
- R. Ishimoto, K. Kamata, K. Suzuki, K. Yamaguchi and N. Mizuno, Dalton Trans., 2015, 44, 10947 RSC.
- S. Pandiaraju, G. Chen, A. Lough and A. K. Yudin, J. Am. Chem. Soc., 2001, 123, 3850 CrossRef CAS PubMed.
- J. Balsells, T. J. Davis, P. Carroll and P. J. Walsh, J. Am. Chem. Soc., 2002, 124, 10336 CrossRef CAS PubMed.
- J. P. Corden, W. Errington, P. Moore, M. G. Partridge and M. G. H. Wallbridge, Dalton Trans., 2004, 1846 RSC.
- B. Schetter, B. Ziemer, G. Schnakenburg and R. Mahrwald, J. Org. Chem., 2008, 73, 813 CrossRef CAS PubMed.
- R. Mahrwald and B. Schetter, Org. Lett., 2006, 8, 281 CrossRef CAS PubMed.
- R. Yu, Y. Yamashita and S. Kobayashi, Adv. Synth. Catal., 2009, 351, 147 CrossRef CAS.
- M. Terada, Y. Matsumoto, Y. Nakamura and K. Mikami, Inorg. Chim. Acta, 1999, 296, 267 CrossRef CAS.
- K. Mikami, M. Ueki, Y. Matsumoto and M. Terada, Chirality, 2001, 13, 541 CrossRef CAS PubMed.
- N. B. McKeown, J. Mater. Chem., 2010, 20, 10588 RSC.
- K. Hong, W. Bak and H. Chun, Inorg. Chem., 2014, 53, 7288 CrossRef CAS PubMed.
- K. Hong and H. Chun, Inorg. Chem., 2013, 52, 9705 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |