
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organisation and ordering of 1D porphyrin polymers synthesised by on-surface Glaser coupling†
Alex
Saywell
*a,
Abigail S.
Browning
a,
Philipp
Rahe
a,
Harry L.
Anderson
 b and
Peter H.
Beton
a
b and
Peter H.
Beton
a
aSchool of Physics & Astronomy, University of Nottingham, Nottingham NG7 2RD, UK. E-mail: alex.saywell@nottingham.ac.uk
bDepartment of Chemistry, University of Oxford, Chemistry Research Laboratory, Oxford OX1 3TA, UK
First published on 20th June 2016
Abstract
One-dimensional polymer chains consisting of π-conjugated porphyrin units are formed via Glaser coupling on a Ag(111) surface. Scanning probe microscopy reveals the covalent structure of the products and their ordering. The conformational flexibility within the chains is investigated via a comparision of room temperature and cryogenic measurements.
The on-surface synthesis of covalently bonded molecular frameworks offers a promising route towards custom 1D and 2D materials.1 Covalent coupling of precursors on a surface potentially offers alternative reaction pathways to those available in solution-phase synthesis via substrate-induced regio- and steroselectivity as well as additional control of the ordering of the resulting polymeric structures. On-surface synthesis is also compatible with characterisation using scanning probe techniques both under ultra-high vacuum (UHV) and liquid conditions2,3 with many previous strategies focusing on Ullman coupling reactions on metallic substrates; in these processes the breaking of a carbon-halogen (C–X) bond is catalytically activated and new C–C bonds are formed.4–9 Alternative strategies (e.g. condensation reactions, dehydration and esterification of boronic acids), which do not include halogen leaving groups that remain on the substrate, have also been studied (see ref. 3 for a recent review). The homo-coupling of terminal alkynes has been studied as an alternative route to covalently bonded frameworks,10,11 with the only by-product being molecular hydrogen, and is utilised within this work.
Interest in many 1D materials relates to their optical and electronic properties, and electronically coupled chains of porphyrins, which possess novel electrical and optical properties,12,13 represent excellent candidates for applications. When synthesised in solution these polymers are too large to be thermally sublimed for study in UHV, even with rapid heating,14 and have to be deposited by alternative techniques, such as electrospray deposition.15–17 Implementation of such techniques has allowed 1D porphyrin oligomers, polymers, and porphyrin rings to be studied.18,19 However, deposition of these molecules often leads to the introduction of disorder in the organisation of adsorbed species. This can occur, for example, through formation of overlapping chains, or partial collapse of more complex supramolecular motifs pre-formed in solution.20,21 It is of interest to explore whether on-surface synthesis of an analogue porphyrin polymer results in similar organisational disorder, or, alternatively, whether direct growth can provide a route to more ordered arrangements.
Here we present on-surface synthesis of 1D polymer chains via homo-coupling of terminal alkyne functionalised porphyrins on a Ag(111) substrate. The resulting polymers are close analogues to materials which have previously been synthesised in solution prior to electrospray deposition on metallic surfaces in vacuum. We observe a high selectivity for 1D Glaser coupling reaction products and find ordered polymer chains with lengths of over 55 nm (>42 porphyrin units). From a combination of room temperature (RT) scanning tunnelling microscopy (STM) and low temperature (LT) STM and frequency-modulated non-contact atomic force microscopy (ncAFM) the covalent nature of the chains, and their internal molecular conformations, are determined and characterised.
The porphyrin monomer unit studied (zinc, [5,15-bis(3,5-di-tert-butylphenyl)-10,20-diethynyl-porphyrin]) is shown in Fig. 1a, with the scheme for the homo-coupling of terminal alkynes given in Fig. 1b. The molecules are deposited, using a ‘rapid heating’ technique, onto a Ag(111) substrate held at RT under UHV conditions, followed by annealing the sample at 120 °C, after which, large, ordered, islands consisting of the polymer chains are observed (Fig. 1c – see ESI† for full experimental details). The islands have an average length of 28 ± 10 nm (≈21 porphyrin units). The periodicity along the chains (indicated by a blue arrow in Fig. 1c – line profile shown in Fig. 1e) is measured to be 1.33 ± 0.07 nm, confirming the covalent nature of the bond; cf. 1.353 nm from crystallographic data22 and 1.33 ± 0.07 nm from studies of similarly covalently bonded porphyrin polymer chains.19Fig. 1d shows a region of close-packed chains. This morphology is very different to the arrangement observed for similar porphyrin polymers deposited on gold by electrospray;19 in that case regions of local parallel alignment of chains were present, but the chain direction was randomised over a length of ∼25 nm and many overlapping chains were observed. Here we identify regions where the polymers exhibit local curvature (see Fig. 1d, dashed line) as expected for these flexible polymers.20,21 Interestingly the hairpin curved region in Fig. 1d appears to form continuous connections between chains in the close-packed regions of the island implying that the length of individual chains is greater (by a factor ∼2) than the island size; therefore average chain length is ≈55 nm (42 porphyrin units) – similar in length to previous solution-synthesised polymers19 but with a much higher degree of structural order.
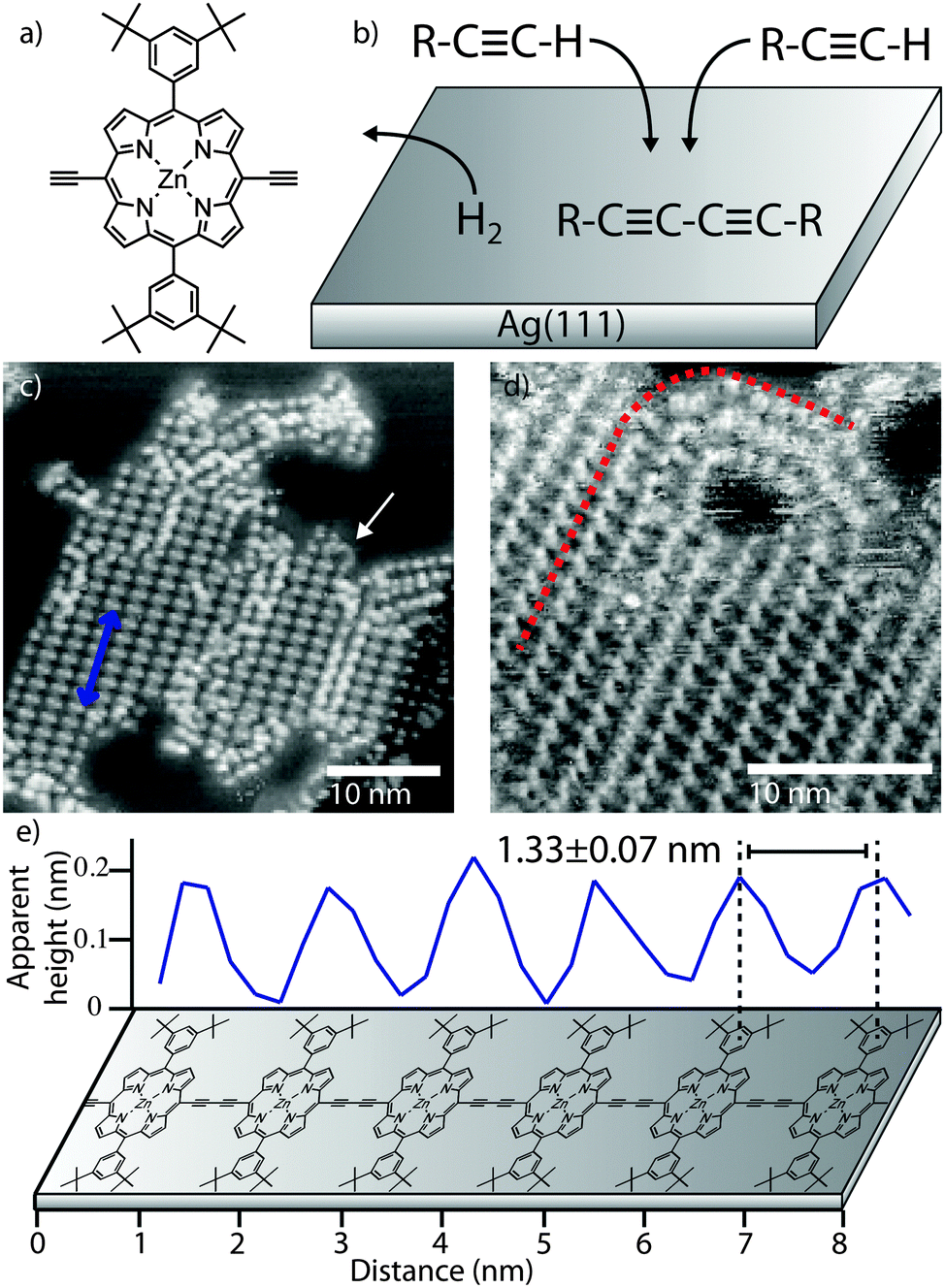 | ||
| Fig. 1 Polymer chains formed on Ag(111) by on-surface synthesis. (a) Chemical structure of porphyrin monomer. (b) Scheme for on-surface reaction. STM images of (c) close-packed 1D polymer islands after deposition and annealing at 120 °C (blue arrow shows row direction and position of line-profile in (e), white arrow shows ‘hair-pin’ bend), (d) close-packed island; linear to curved transition indicated. (e) Line-profile acquired from (c) with dimensions and polymer model. Image parameters: tunnel current, I = 30 pA, sample voltage, V = −1.80 V. | ||
Previous studies of 1D polymerisation via Glaser coupling have noted: (1) low mass monomers desorb before the reaction could be initiated, and (2) the formation of nonlinear en-yne links and 2D branched structures instead of the targeted 1D chains.11 Here we observe 1D butadiyne-linked products without co-existing 2D structures produced by branching. This is attributed to the adsorption energy of the monomers being sufficient to suppress molecular desorption during annealing, and the surface confinement and geometry of the molecule driving the reaction towards the 1D product.
In order to characterise the internal structure of the polymer chains STM measurements were acquired at LT (78 K).‡Fig. 2a shows an STM image of an island of porphyrin chains with an arrangement of ‘bright’ and ‘dark’ features, with high resolution images revealing the sub-molecular structure of the porphyrin units (Fig. 2b – pairs of features correspond to a single molecule). From the dimensions of the lattice of the porphyrin cores [A = 1.35 ± 0.07 nm, B = 2.00 ± 0.10 nm, θ = 80 ± 1°] and separation of the sub-molecular features [C = D = 0.82 ± 0.04 nm] we assign the sub-molecular features to individual tertiary-butyl (t-Bu) side-groups of the monomers (Fig. 2c–e). It is particularly interesting to note that the angle, β, formed between the line joining two t-Bu groups of a monomer and the covalently bonded row direction of the polymer (Fig. 2c) is different for the molecules which appear with a brighter and darker contrast in the STM images as shown in Fig. 2b. The measured angles of ∼90°, β2, and ∼60°, β1, respectively for ‘bright’ and ‘dark’ molecules, correspond well to the expected angle formed by the di-t-butyl-phenyl side-groups of the porphyrin core being in one of two conformations (shown in Fig. 2d). In the case where the aryl ring of the side-groups is rotated by 90° relative to the surface plane (as previously observed23,24), the ‘upright’ conformation, β is expected to be 90°. Whereas in the ‘tilted’ conformation the aryl ring is only slightly rotated from the surface plane (as previously reported for related porphyrin species25) and an opposite tilting of the rings on opposing sides of the porphyrin core would give rise to the t-Bu groups producing β ≈ 60–70°, as observed in experiment.
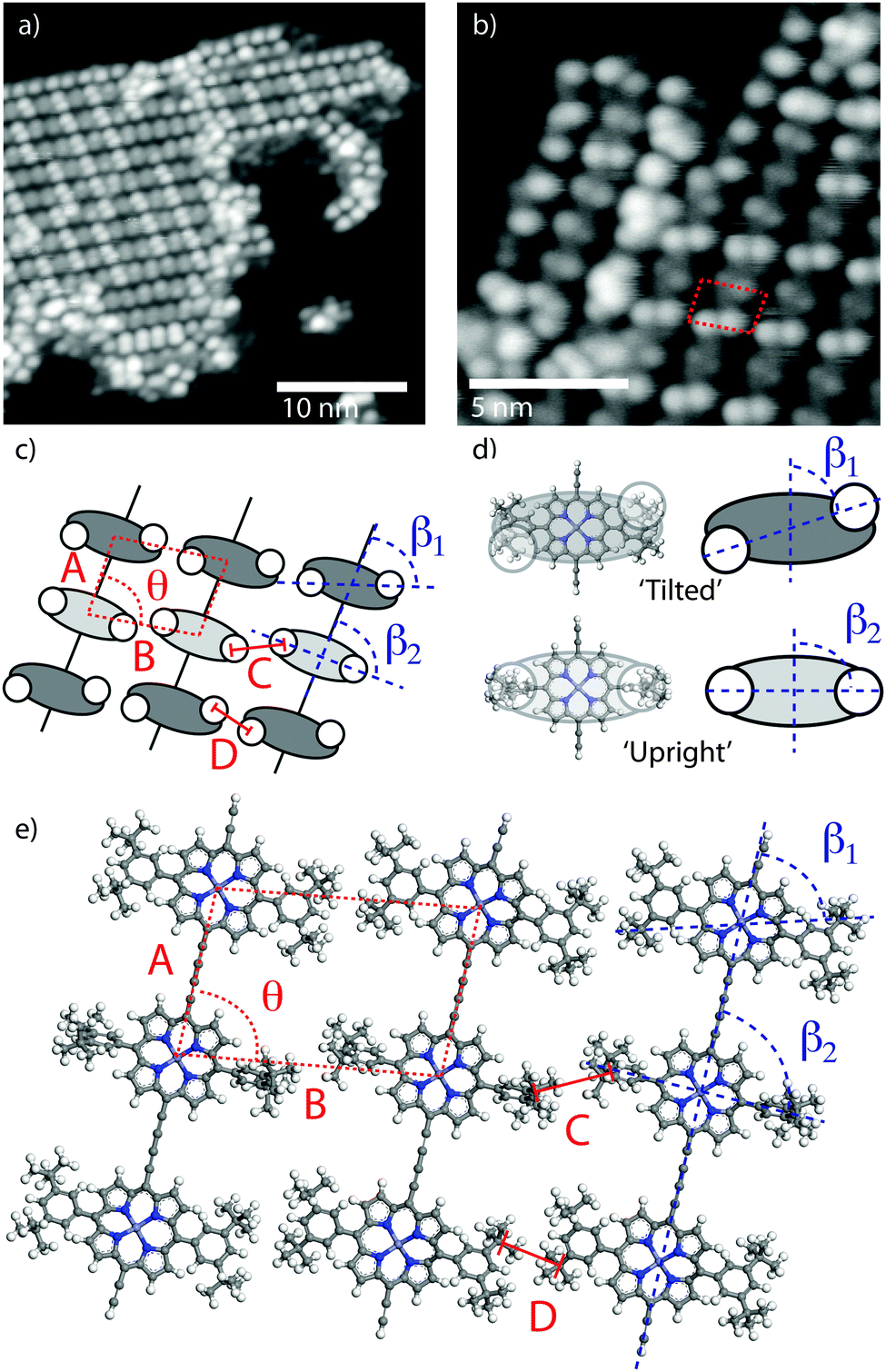 | ||
| Fig. 2 Characterisation of 1D polymer chains by LT STM. STM images showing (a) structure of close-packed islands, and (b) sub-molecular resolution; porphyrin lattice indicated. (c) Scheme showing observed sub-molecular features and porphyrin lattice. (d) Molecular structure and schematics for the two conformers. (e) Molecular model of the close-packed islands formed from polymer chains. Image parameters: (a) tunnel current, I = 10 pA, sample voltage V = −1.0 V, (c) tunnel current, I = 2 pA, sample voltage, V = −1.5 V. Temperature, T = 78 K. | ||
From the experimentally acquired unit cell dimensions and assigned conformations of the porphyrin side-groups a structural model for the close-packed porphyrin islands is elucidated (Fig. 2e). The model is in good agreement with the experimentally observed structure of alternating ‘bright’ and ‘dark’ molecules (due to the molecular conformations). Conformational flexibility has been discussed in the context of surface confined polymer chains, where interactions between adjacent monomers and between monomers and the substrate drives the system into a periodic arrangement.26 Here, conformational ordering appears to be driven by interactions between neighbouring polymers, rather than within the molecular chain, and is stabilised by interdigitation of t-Bu groups across neighbouring rows (Fig. 2eC and D). Interdigitation patterns have been observed for the stabilisation of related porphyrin molecules functionalised with linear alkyl chains,19 and have also been seen to drive the structures formed by large 3D molecular species.17 Here interdigitation of t-Bu groups from neighbouring chains stabilises the observed periodic structure and gives rise to a more ordered arrangement than for solution-synthesised porphyrin polymers deposited onto a substrate in UHV.
It is known that STM provides information about the local density of states (LDOS), and hence STM images do not necessarily represent topographical heights of molecule–substrate systems. Complementary information can be obtained from ncAFM measurements, where sub-molecular contrast arises from short-range forces.27 In this mode, data may be more readily interpreted in terms of a topographic height.28Fig. 3a shows an STM image (constant current mode) of a close-packed island of 1D polymers with bright and dark features clearly visible (highlighted with red and blue ellipses, respectively). Constant height ncAFM images acquired in the same region show that as tip-sample separation is decreased the measured frequency shift, df, above the sub-molecular features initially becomes negative – indicative of an attractive interaction (Fig. 3b, dark contrast).27 Further decrease in tip-sample separation shows a shift in df towards positive values above the sub-molecular features, indicating an increase in the repulsive contribution of the interaction. This effect is particularly pronounced above sub-molecular features imaged as brighter protrusions in STM (Fig. 3c, red ellipse). Consequently, the ncAFM data indicates that the tip is being brought into close proximity with the t-Bu groups at these positions. Therefore the features imaged as bright protrusions in STM (red ellipse) are indeed topographically higher features compared to those in the blue ellipse, in full agreement with our model of ‘upright’ and ‘tilted’ conformations.
 | ||
| Fig. 3 Characterisation of islands and curved chains by ncAFM. (a) STM image of a close-packed island. (b and c) Same area as in (a) measured with ncAFM; tip-sample separation is decreased by 0.05 nm from (b) to (c). (d) STM image of a curved chain. (e and f) ncAFM images of a curved chain; the tip-sample separation is decreased by 0.05 nm from (e) to (f). Image parameters: (a and d) STM – tunnel current, I = 10 pA, sample voltage, V = −1.8 V, (b, c, e and f) ncAFM – constant height, sample voltage, (b and c) V = +0.01 V, (e and f) V = 0 V. Temperature, T = 78 K. | ||
Fig. 3d–f shows a similar series of images for a curved polymer chain. As this chain is not stabilised within an island the monomers do not exhibit the periodic structure. Discontinuities in the df image shown in Fig. 3f (black arrows) point to tip-induced molecular changes with decreasing tip-sample separation (i.e. interaction between the tip and molecule). Further decrease in the tip-sample separation results in manipulation of the polymer chain (see ESI†).
We may compare the conformations observed at LT with those of the RT assemblies. At RT the value of β for the close-packed chains is 90°, indicating that all molecules exhibit the ‘upright’ conformation. Within the islands there are areas of high contrast (white arrows Fig. 4a) corresponding to domain boundaries – porphyrin units being misaligned between neighbouring rows (red line in Fig. 4a). The domain boundaries can be modelled by the interdigitation of ‘upright’ t-Bu groups in neighbouring rows, and leads to the observed misalignment across the polymer chains (proposed model in Fig. 4b). The fact the domain boundaries ‘lock-in’ the ‘upright’ structure of the porphyrin suggests that we do not see a time average effect, where the molecules oscillate between two equivalent ‘tilted’ conformations on a timescale faster than the STM image acquisition, but instead observe a distinct conformation.
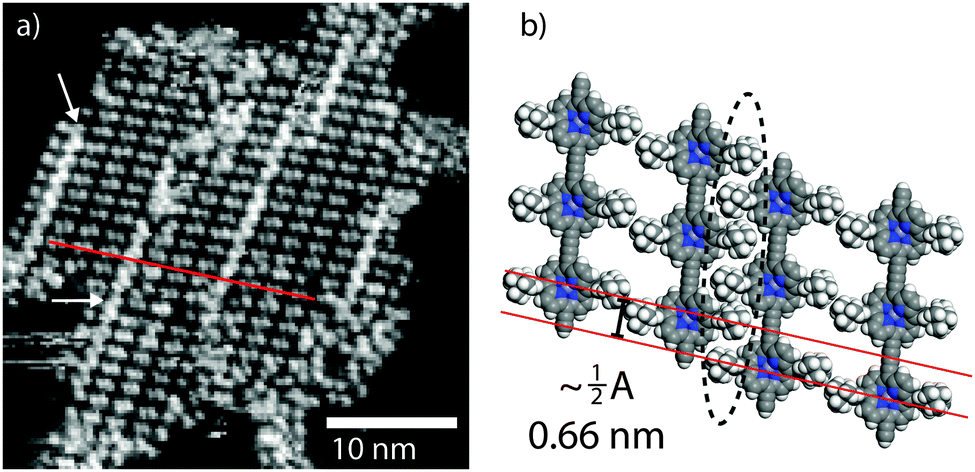 | ||
| Fig. 4 RT STM images of the molecular packing. (a) STM image of a close-packed island showing the domain boundaries present (indicated with white arrows). (b) Molecular model of the domain boundary; domain boundary highlighted by dashed ellipse. Image parameters: tunnel current, I = 50 pA, sample voltage, V = −1.50 V. | ||
In conclusion, we have demonstrated the on-surface synthesis of 1D polymer structures consisting of porphyrin sub-units via Glaser coupling on Ag(111). The majority of on-surface reactions to date have focused on relatively planar molecules with little or no conformational flexibility. However, for structures containing large, flexible, molecules the effect of molecular conformations on the stability of the assemblies needs to be considered. From characterisation of the structures we have assigned sub-molecular features to two conformations, ‘tilted’ and ‘upright’; at RT an all ‘upright’ arrangement is observed while at low temperature (78 K) a periodic structure is formed. The on-surface synthesis of these polymers leads to much more ordered assemblies than those formed by solution-synthesis and subsequent deposition. This finding highlights the importance of on-surface synthesis as a method to produce novel 1D systems for study in UHV and that conformational flexibility plays an important role in the stabilisation of the surface confined structures.
A. S. and P. R. would like to acknowledge funding from the People Programme (Marie Curie Actions) of the European Union's Seventh Framework Programme (623992-TOPCHEM and 628439-DECiMOL, respectively). We thank the U. K Engineering and Physical Sciences Research Council for support through grant EP/J006939/1, and thank Ioannis Lekkas (University of Nottingham) and Franz Giessibl and his group at the University of Regensburg for help and advice in relation to qPlus sensor construction. The experimental data on which this work is based, including STM and ncAFM image files, may be found at http://dx.doi.org/10.17639/nott.47.
Notes and references
- M. El Garah, J. M. MacLeod and F. Rosei, Surf. Sci., 2013, 613, 6–14 CrossRef CAS.
- L. Dong, P. N. Liu and N. Lin, Acc. Chem. Res., 2015, 48, 2765–2774 CrossRef CAS PubMed.
- R. Lindner and A. Kühnle, ChemPhysChem, 2015, 16, 1582–1592 CrossRef CAS PubMed.
- L. Grill, M. Dyer, L. Lafferentz, M. Persson, M. V. Peters and S. Hecht, Nat. Nanotechnol., 2007, 2, 687–691 CrossRef CAS PubMed.
- J. Cai, P. Ruffieux, R. Jaafar, M. Bieri, T. Braun, S. Blankenburg, M. Muoth, A. P. Seitsonen, M. Saleh, X. Feng, K. Müllen and R. Fasel, Nature, 2010, 466, 470–473 CrossRef CAS PubMed.
- R. Gutzler, H. Walch, G. Eder, S. Kloft, W. M. Heckl and M. Lackinger, Chem. Commun., 2009, 4456 RSC.
- J. A. Lipton-Duffin, O. Ivasenko, D. F. Perepichka and F. Rosei, Small, 2009, 5, 592–597 CrossRef CAS PubMed.
- J. C. Russell, M. O. Blunt, J. M. Garfitt, D. J. Scurr, M. Alexander, N. R. Champness and P. H. Beton, J. Am. Chem. Soc., 2011, 133, 4220–4223 CrossRef CAS PubMed.
- G. Eder, E. F. Smith, I. Cebula, W. M. Heckl, P. H. Beton and M. Lackinger, ACS Nano, 2013, 7, 3014–3021 CrossRef CAS PubMed.
- Y.-Q. Zhang, N. Kepčija, M. Kleinschrodt, K. Diller, S. Fischer, A. C. Papageorgiou, F. Allegretti, J. Björk, S. Klyatskaya, F. Klappenberger, M. Ruben and J. V. Barth, Nat. Commun., 2012, 3, 1286 CrossRef PubMed.
- H.-Y. Gao, H. Wagner, D. Zhong, J.-H. Franke, A. Studer and H. Fuchs, Angew. Chem., Int. Ed., 2013, 52, 4024–4028 CrossRef CAS PubMed.
- H. L. Anderson, S. J. Martin and D. D. C. Bradley, Angew. Chem., Int. Ed. Engl., 1994, 33, 655–657 CrossRef.
- G. Sedghi, V. M. GarcíaSuárez, L. J. Esdaile, H. L. Anderson, C. J. Lambert, S. Martín, D. Bethell, S. J. Higgins, M. Elliott, N. Bennett, J. E. Macdonald and R. J. Nichols, Nat. Nanotechnol., 2011, 6, 517–523 CrossRef CAS PubMed.
- G. Rapenne, L. Grill, T. Zambelli, S. M. Stojkovic, F. Ample, F. Moresco and C. Joachim, Chem. Phys. Lett., 2006, 431, 219–222 CrossRef CAS.
- H. Tanaka and T. Kawai, Nat. Nanotechnol., 2009, 4, 518–522 CrossRef CAS PubMed.
- S. Rauschenbach, R. Vogelgesang, N. Malinowski, J. W. Gerlach, M. Benyoucef, G. Costantini, Z. Deng, N. Thontasen and K. Kern, ACS Nano, 2009, 3, 2901–2910 CrossRef CAS PubMed.
- A. Saywell, G. Magnano, C. J. Satterley, L. M. A. Perdigão, A. J. Britton, N. Taleb, M. d. C. Giménez-López, N. R. Champness, J. N. O'Shea and P. H. Beton, Nat. Commun., 2010, 1, 75 Search PubMed.
- M. C. O'Sullivan, J. K. Sprafke, D. V. Kondratuk, C. Rinfray, T. D. W. Claridge, A. Saywell, M. O. Blunt, J. N. O'Shea, P. H. Beton, M. Malfois and H. L. Anderson, Nature, 2011, 469, 72–75 CrossRef PubMed.
- A. Saywell, J. K. Sprafke, L. J. Esdaile, A. J. Britton, A. Rienzo, H. L. Anderson, J. N. O'Shea and P. H. Beton, Angew. Chem., Int. Ed., 2010, 49, 9136–9139 CrossRef CAS PubMed.
- S. A. Svatek, L. M. A. Perdigão, A. Stannard, M. B. Wieland, D. V. Kondratuk, H. L. Anderson, J. N. O'Shea and P. H. Beton, Nano Lett., 2013, 13, 3391–3395 CrossRef CAS PubMed.
- D. V. Kondratuk, L. M. A. Perdigão, A. M. S. Esmail, J. N. O'Shea, P. H. Beton and H. L. Anderson, Nat. Chem., 2015, 7, 317–322 CrossRef CAS PubMed.
- P. N. Taylor, J. Huuskonen, G. Rumbles, R. T. Aplin, E. Williams and H. L. Anderson, Chem. Commun., 1998, 909–910 RSC.
- T. A. Jung, R. R. Schlittler and J. K. Gimzewski, Nature, 1997, 386, 696–698 CrossRef CAS.
- F. Buchner, K. Comanici, N. Jux, H.-P. Steinrück and H. Marbach, J. Phys. Chem. C, 2007, 111, 13531–13538 CrossRef CAS.
- D. Heim, D. Écija, K. Seufert, W. Auwärter, C. Aurisicchio, C. Fabbro, D. Bonifazi and J. V. Barth, J. Am. Chem. Soc., 2010, 132, 6783–6790 CrossRef CAS PubMed.
- A. Saywell, W. Greń, G. Franc, A. Gourdon, X. Bouju and L. Grill, J. Phys. Chem. C, 2014, 118, 1719–1728 CrossRef CAS.
- L. Gross, F. Mohn, N. Moll, P. Liljeroth and G. Meyer, Science, 2009, 325, 1110–1114 CrossRef CAS PubMed.
- B. Schuler, W. Liu, A. Tkatchenko, N. Moll, G. Meyer, A. Mistry, D. Fox and L. Gross, Phys. Rev. Lett., 2013, 111, 106103 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental methods and additional experimental information. See DOI: 10.1039/c6cc03758e |
| ‡ Transfer between RT and LT UHV systems facilitated via a ‘vacuum suitcase’ transfer system. |
| This journal is © The Royal Society of Chemistry 2016 |