
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Non-coordinating anions assemble cyanine amphiphiles into ultra-small fluorescent nanoparticles†
Ievgen
Shulov
ab,
Youri
Arntz
a,
Yves
Mély
a,
Vasyl G.
Pivovarenko
b and
Andrey S.
Klymchenko
*a
aLaboratoire de Biophotonique et Pharmacologie, UMR 7213 CNRS, Université de Strasbourg, Faculté de Pharmacie, 74, Route du Rhin, 67401 ILLKIRCH Cedex, France. E-mail: andrey.klymchenko@unistra.fr; Tel: +33 368 85 42 55
bOrganic Chemistry Department, Chemistry Faculty, Taras Shevchenko National University of Kyiv, 01601 Kyiv, Ukraine
First published on 23rd May 2016
Abstract
A non-coordinating anion, fluorinated tetraphenylborate, assembles specially designed cationic cyanine amphiphiles into 7–8 nm fluorescent nanoparticles that are >40-fold brighter than a single cyanine dye. This kind of anion, combining hydrophobic and electrostatic forces in aqueous media, constitutes promising building blocks in the self-assembly of functional nanomaterials.
Molecular self-assembly is a powerful bottom-up approach to generate a large variety of nanoscopic and mesoscopic functional materials.1–3 However, self-assembly exploits a quite limited number of non-covalent interactions:4 (1) H-bonding and (2) metal-coordination, featuring high directionality, (3) hydrophobic and (4) pi-stacking interactions, which are especially strong in aqueous media, (5) electrostatic interactions, and (6) van der Waals interactions. A successful bottom-up assembly in aqueous media requires a combination of several types of these interactions, as is successfully realized in the preparation of supramolecular nanoparticles, (NPs),3,5 fibers and tubes.1,6 A combination of electrostatic and hydrophobic interactions is a fruitful approach, because the former is very efficient in apolar media. Therefore, ionic self-assembly of functional nanostructures7 exploits surfactants bearing charged and hydrophobic moieties to assemble charged dyes, polyelectrolytes, etc.8 A promising direction is to use non-coordinating anions, which are highly hydrophobic and weakly hydrated ions.9 The key representatives are tetraphenylborate (TPB) anions bearing hydrophobic aromatic groups. Their fluorinated analogues are particularly interesting due to the super-hydrophobic nature of fluorous compounds10 and are successfully used in phase transfer catalysis and as charge carriers inside electrodes.9 Our recent studies showed that tetraphenylborates can help in encapsulation of organic dyes inside polymer11 and lipid12 matrices of nanocarriers. Importantly, in the presence of non-coordinating anions, cationic dyes can form nanoparticles, called ion-associated NPs13,14 or GUMBOS.15 Fluorination of these counterions is crucial to achieve high fluorescence quantum yield of NPs and sufficient stability in biological media.16 In all these examples formation of NPs is kinetically controlled, because relatively hydrophobic cationic dyes and non-coordinating anions give highly hydrophobic salts that rapidly precipitate in aqueous media. However, so far these non-coordinating hydrophobic counterions have never been used to induce assembly of amphiphiles into micelles, which is a fundamental, thermodynamically controlled process giving ultra-small NPs.17 Assembly of organic dyes into micelles is particularly interesting because it enables preparation of fluorescent NPs,18 which have enormous potential for bioimaging applications.19 However, concentrating dyes at the nanoscale leads to aggregation caused quenching (ACQ),20 so that the reported examples of ultra-small micelles from organic dye amphiphiles display relatively low fluorescence quantum yields. In this respect, fluorinated counterions that can prevent cationic dyes from ACQ16 are of particular importance to generate bright fluorescent micellar NPs.
In the present work, we show that non-coordinating counterions can trigger the assembly of amphiphilic cyanine-3 and cyanine-5 dyes into ultra-small fluorescent nanoparticles. We selected cyanines, because they are cationic dyes with a positive charge delocalized within a large hydrophobic aromatic structure. This should allow combining hydrophobic and electrostatic interactions for assembly with the non-coordinating anions. Moreover, these dyes are bright owing to high molar absorption coefficients and good fluorescence quantum yields. We prepared symmetric gemini-like amphiphiles containing two hydrophilic PEG groups and two apolar hydrocarbon chains (Fig. 1). This design should enable the preparation of micelles where cyanine dyes are oriented parallel to the micelle surface with freedom to form emissive J- or non-emissive H-aggregates, well known for cyanines.21 The final Cy3A and Cy5A amphiphiles were synthesized in multiple steps, where Cy3 and Cy5 derivatives bearing two carboxylic groups were the corresponding intermediates (Scheme S1, ESI†).
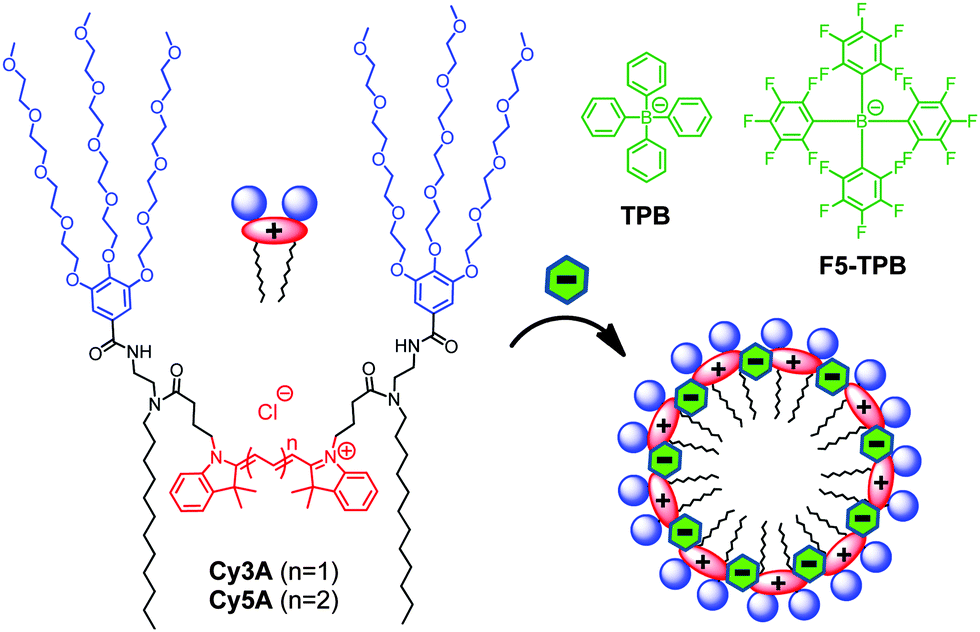 | ||
| Fig. 1 Cyanine amphiphiles and their expected assembly into micelles in the presence of non-coordinating anions. | ||
First, we studied the capacity of Cy-amphiphiles to assemble in aqueous media using absorption spectroscopy (Fig. 2). At increasing concentrations of Cy3A and Cy5A in water, the contribution of the short-wavelength shoulder with respect to the peak maximum Ash/Amax was found to increase, whereas in methanol this ratio was constant (Fig. S1 and S2, ESI†). This increased shoulder suggests that both dyes at higher concentrations (1–5 μM) may form H-aggregates, characterized by short-wavelength absorption.13,21 The addition of non-coordinating anions TPB and F5-TPB increased the Ash/Amax ratio in the absorption spectra of Cy3A and Cy5A, shifting the whole absorption band slightly to red (Fig. 2 and Fig. S2, ESI†). In the case of Cy3A, Ash/Amax increased gradually with anion concentration (Fig. 2B), reaching saturation at an anion/dye ratio of 1–2, so that counterions induced assembly of Cy3A at an equimolar ratio. A similar, but less pronounced effect was observed for Cy5A (data not provided). At 10 mole excess of TPB and F5-TPB anions, Ash/Amax remained stably high for Cy3A/Cy5A concentrations >0.5 μM (Fig. S1 and S2, ESI†), indicating that with these counterions the assembly took place below 0.5 μM. The Ash/Amax ratio for TPB was higher than for F5-TPB, showing some differences in the dye assembly.
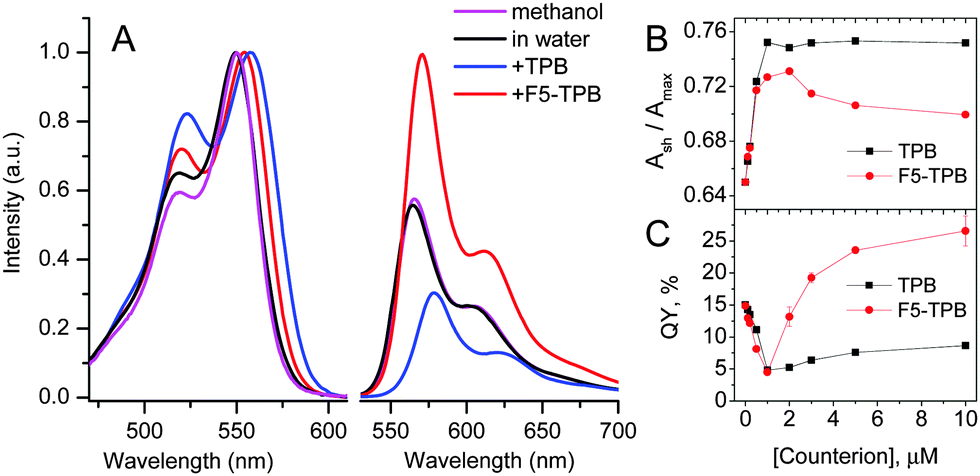 | ||
| Fig. 2 (A) Absorption (normalized) and fluorescence spectra of 1 μM solutions of Cy3A in methanol and water in the presence of TPB or F5-TPB (10 mol. eq.). Emission spectra were normalized to the same absorbance at 520 nm. λex = 520 nm. (B) The ratio of the absorption bands Ash/Amax and (C) the fluorescence quantum yield of 1 μM Cy3A in water vs. counterion concentration. | ||
The most interesting effects were observed in fluorescence (Fig. 2B and Fig. S2, ESI†). At a 1/1 counterion/Cy3A molar ratio the emission maximum shifted to red (Fig. S3, ESI†), while the fluorescence quantum yield (QY) decreased strongly for both TPB and F5-TPB (Fig. 2C), indicating an aggregation-caused quenching. A further increase in the TPB concentration produced a small increase in QY, whereas for F5-TPB it increased many-fold (Fig. 2C). At 10 mole excess of F5-TPB, the QY values were larger than in water reaching 25 and 8% for Cy3A and Cy5A, respectively, and their emission maxima were red shifted (Table 1). To confirm that this efficient fluorescence is observed from self-assembled nanostructures, we performed fluorescence anisotropy measurements. In dye assemblies, the migration of energy (exciton diffusion) can cause a strong decrease of fluorescence anisotropy as the energy hops within dyes of different orientations.22 In the absence of non-coordinating anions, our fluorescent amphiphiles showed an anisotropy value in water close to that in methanol, thus reflecting the rotation of free dye molecules (Table 1). Remarkably, in the presence of F5-TPB the anisotropy decreased 5–8 fold for both dyes, confirming that the counterions induced their assembly into emissive multi-fluorophore nanostructures.
| Sample | λ abs (nm) | λ em (nm) | FWHM (nm) | QY (%) | Anisotropy |
|---|---|---|---|---|---|
| a FWHM – full width at half maximum; QY – fluorescence quantum yield measured using reference dyes (ESI). | |||||
| Cy3A/MeOH | 550 | 566 | 40 | 15 | 0.153 |
| Cy3A | 550 | 565 | 57 | 15 | 0.171 |
| Cy3A/F5-TPB | 555 | 572 | 59 | 25 | <0.02 |
| Cy3A/TPB | 557 | 581 | 66 | 8 | 0.034 |
| Cy5A/MeOH | 646 | 666 | 40 | 39 | 0.115 |
| Cy5A | 647 | 667 | 44 | 4 | 0.140 |
| Cy5A/F5-TPB | 651 | 687 | 39 | 8 | <0.02 |
| Cy5A/TPB | 655 | 697 | 46 | 1 | 0.017 |
Dynamic light scattering (DLS) measurements suggested that Cy3A in the presence of 10-fold excess of TPB and F5-TPB formed particles of 7 ± 1 nm (polydispersity index, PDI = 0.18) and 12 ± 3 nm (PDI = 0.22) hydrodynamic diameter, respectively. Remarkably, the zeta potential of Cy3A/F5-TPB NPs was strongly negative (−80 ± 15 mV), likely due to adsorption at their surface of F5-TPB counterions, present in the excess. At a 1/1 counterion/Cy3A ratio, the particle size was larger for both counterions (∼20 nm), likely due to aggregation of neutral NPs, in line with the decrease in the QY values at this ratio (Fig. 2C). Cy5A samples were not measured, because of their direct excitation by the DLS laser source. Then, our NPs were studied by Atomic Force Microscopy (AFM). The dye amphiphiles in the presence of TPB or F5-TPB counterions were deposited on mica in an aqueous solution of calcium chloride to bridge the negatively charged NPs with the negatively charged glass surface. The best images were obtained for Cy3A with F5-TPB counterions, where relatively homogeneous spherical NPs of 7 nm height were observed (Fig. 3), in line with the DLS data suggesting formation of ultra-small NPs. Taking into account that the length of the Cy3A amphiphile (from the end of the alkyl chain to the end of the PEG chain) is about ∼3 nm, this small size and spherical shape suggest a micellar organization of NPs. In these micelles, the alkyl chains, the dye and the counterions form the core, while the PEG groups play the role of a polar shell exposed to water (Fig. 1). Other amphiphile/counterion combinations gave similar sizes (Fig. 3C), though the particles appeared to be more heterogeneous and larger in the XY directions (Fig. S4 and S5, ESI†), which could be related to their aggregation at the mica surface.
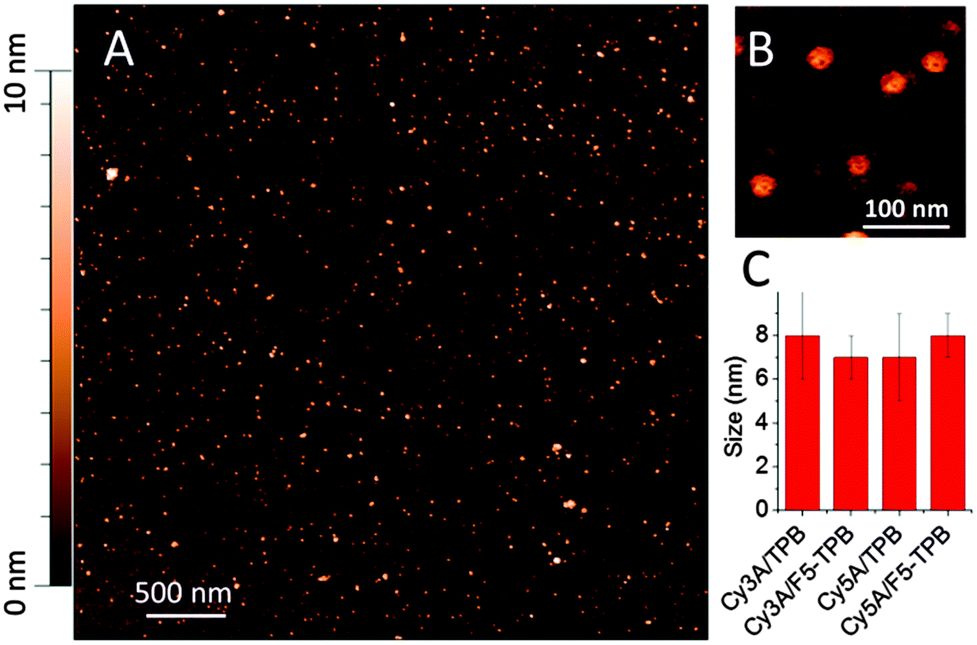 | ||
| Fig. 3 Atomic force microscopy of NPs formed by Cy3A and F5-TPB (10 mol. eq.) (A), a zoomed image (B) and particle sizes measured by AFM (height) for different samples (C). | ||
Then, to evaluate their single particle properties, we performed fluorescence correlation spectroscopy (FCS). This unique technique, which measures diffusion of emissive species through a focal volume, provides direct access to the size, concentration and brightness of NPs.23,24 As we used a two-photon excitation source, we were able to study only Cy3A samples, which present an appreciable two-photon absorption cross-section at 760 nm.25 Only solutions of Cy3A with F5-TPB displayed a stable FCS signal, which could be measured and analyzed. FCS data suggested that the size of NPs was 6.5 nm (Table S1, ESI†), in perfect line with the AFM data (Fig. 3B). Taking together, all the used techniques suggest an ultra-small size of Cy3A/F5-TPB NPs, confirming our hypothesis on their micellar organization. Moreover, the brightness of these particles is close to 6 molecules of tetramethyl-rhodamine (TMR). Taking into account the two-photon absorption cross-section (σ2) of rhodamine at 760 nm (85 GM) and its quantum yield in water (0.4) and those of the Cy3A dye (25 GM and 0.2, respectively),25 we calculated that 6 rhodamines are equivalent to 41 Cy3A dyes. This calculation suggests that the micelle contains around 41 dyes, which is a typical aggregation number of amphiphiles (25–100). Finally, from the concentration of emissive species measured by FCS, and the total Cy3A concentration, we could find that the number of dyes per micelle is 25, in agreement with the number obtained from the single particle brightness.
To generate Forster Resonance Energy Transfer (FRET) inside our micelles, we prepared them from the mixtures of Cy3A (donor) and Cy5A (acceptor) in the presence of F5-TPB. Increasing the acceptor concentration (i.e. a decrease in the donor/acceptor ratio) resulted in an increase of the acceptor emission and a decrease in the donor emission (Fig. 4A), a typical signature of FRET. The relative FRET efficiency (IA/(ID + IA) × 100) increased with a decrease in the donor/acceptor ratios up to 20/1 (Fig. 4B). Below this ratio, the relative FRET efficiency remained stable around 80%. Probably, at donor/acceptor ratios ≤20/1 each micelle contained at least one acceptor, in line with the aggregation number >20 estimated by FCS. Moreover, at a donor/acceptor ratio ≤20/1, we cannot exclude a partial donor/acceptor phase separation, described for other dye NPs.26 Due to the efficient FRET from multiple donors to a single acceptor, our micelles behave like a light-harvesting system. The antenna effect (AE) of our micelles, calculated as the ratio of intensities of donor and acceptor dyes in the excitation spectra, reached a value of 30 (Fig. 4C). Based on the expression of the antenna effect (see the ESI†) and assuming one acceptor per particle for high donor/acceptor ratios, the estimated number of donors per particle is 63. The latter value is close to the aggregation number estimated by other methods (41 and 23), though some discrepancies are clearly related to the limitations of the methods and the assumptions made.
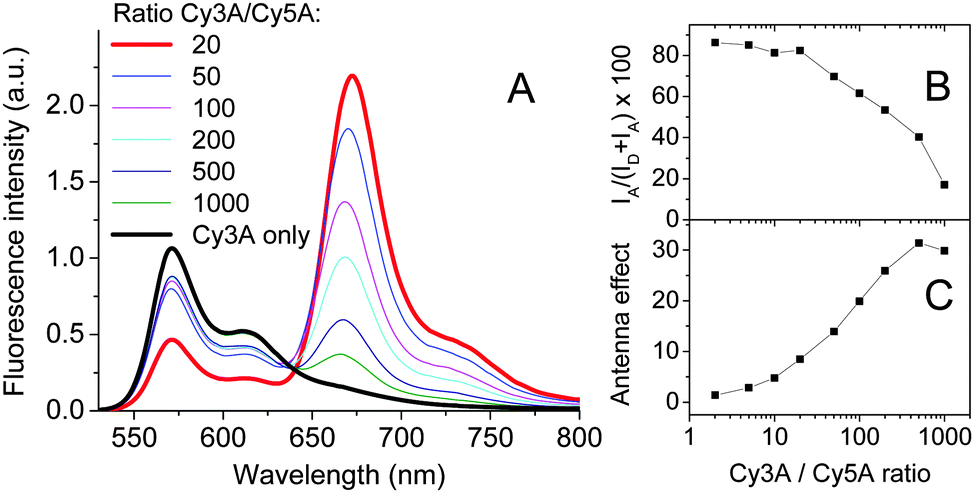 | ||
| Fig. 4 Fluorescence spectra showing FRET from Cy3A to Cy5A co-assembled with F5-TPB (10 mol. eq.) at different donor/acceptor ratios (A). The relative FRET efficiency (B) and antenna effect (C) as a function of the donor/acceptor ratio. Concentrations of Cy3A was systematically 1 μM. λex = 520 nm. | ||
As FRET is sensitive to the donor–acceptor distance, it can be used to study the integrity of nanostructures.24,27 Remarkably, dilution up to 1000-fold of our FRET NPs originally prepared at 1 μM concentration produced only minor modifications in their dual emission (Fig. S6, ESI†), indicating that NPs remained nearly intact. Therefore, in the presence of F5-TPB the critical micellar concentration of Cy3A should be <1 nM, whereas Cy3A alone does not aggregate well even at 1 μM (Fig. S1, ESI†). This result shows that charged amphiphiles having poor capacity to self-assemble could be brought together into stable nanostructures by non-coordinating anions.
Finally, we verified whether the counterion-assembled cyanine micelles were stable over time by following the absorbance values of these solutions in plastic cuvettes (Fig. S7, ESI†). For both Cy3A and Cy5A with chloride and TPB anions, the absorbance clearly decreased over time, suggesting sedimentation of the particles and their adsorption on the walls of the cuvette. In sharp contrast, both Cy3A and Cy5A in the presence of highly fluorinated F5-TPB anions displayed almost negligible decrease in the absorbance, indicating a much higher stability of the obtained micelles.
In conclusion, using a non-coordinating anion, fluorinated tetraphenylborate, specially designed amphiphilic cyanine dyes were assembled into ∼7–8 nm fluorescent NPs with minimized self-quenching. The obtained cyanine-3-based micelles were >40-fold brighter than the single cyanine dye. They exhibited efficient FRET to a single acceptor as well as stability to dilution up to 1 nM of the amphiphiles. The non-coordinating anions function as a glue and a spacer of dye amphiphiles, leading to small, stable and highly emissive fluorescent NPs. Thus, non-coordinating anions, especially fluorinated analogues, provide superior control for the assembly of charged dye amphiphiles into functional nanomaterials.
I. S. acknowledges support from French Embassy. This work was supported by ERC Consolidator grant BrightSens 648528.
Notes and references
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- K. Ariga, J. P. Hill, M. V. Lee, A. Vinu, R. Charvet and S. Acharya, Sci. Technol. Adv. Mater., 2008, 9, 014109 CrossRef CAS; S. Furukawa, J. Reboul, S. Diring, K. Sumida and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5700–5734 RSC.
- C. Stoffelen and J. Huskens, Small, 2016, 12, 96–119 CrossRef CAS PubMed.
- J. M. Lehn, Science, 2002, 295, 2400–2403 CrossRef CAS PubMed.
- L. Graña Suárez, W. Verboom and J. Huskens, Chem. Commun., 2014, 50, 7280–7282 RSC.
- T. Shimizu, M. Masuda and H. Minamikawa, Chem. Rev., 2005, 105, 1401–1443 CrossRef CAS PubMed; J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef PubMed.
- C. F. J. Faul, Acc. Chem. Res., 2014, 47, 3428–3438 CrossRef CAS PubMed; C. F. J. Faul and M. Antonietti, Adv. Mater., 2003, 15, 673–683 CrossRef.
- R. Ahmed, S. K. Patra, I. W. Hamley, I. Manners and C. F. J. Faul, J. Am. Chem. Soc., 2013, 135, 2455–2458 CrossRef CAS PubMed; Y. Huang, Y. Yan, B. M. Smarsly, Z. Wei and C. F. J. Faul, J. Mater. Chem., 2009, 19, 2356–2362 RSC; Y. Guan, M. Antonietti and C. F. J. Faul, Langmuir, 2002, 18, 5939–5945 CrossRef; M. Antonietti and C. Goltner, Angew. Chem., Int. Ed., 1997, 36, 910–928 CrossRef.
- I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2004, 43, 2066–2090 CrossRef CAS PubMed.
- M. Cametti, B. Crousse, P. Metrangolo, R. Milani and G. Resnati, Chem. Soc. Rev., 2012, 41, 31–42 RSC.
- A. Reisch, P. Didier, L. Richert, S. Oncul, Y. Arntz, Y. Mely and A. S. Klymchenko, Nat. Commun., 2014, 5, 4089 CAS.
- V. N. Kilin, H. Anton, N. Anton, E. Steed, J. Vermot, T. E. Vandamme, Y. Mely and A. S. Klymchenko, Biomaterials, 2014, 35, 4950–4957 CrossRef CAS PubMed.
- H. Yao and K. Ashiba, RSC Adv., 2011, 1, 834–838 RSC.
- H. Yao, M. Yamashita and K. Kimura, Langmuir, 2009, 25, 1131–1137 CrossRef CAS PubMed.
- D. K. Bwambok, B. El-Zahab, S. K. Challa, M. Li, L. Chandler, G. A. Baker and I. M. Warner, ACS Nano, 2009, 3, 3854–3860 CrossRef CAS PubMed; A. N. Jordan, S. Das, N. Siraj, S. L. de Rooy, M. Li, B. El-Zahab, L. Chandler, G. A. Baker and I. M. Warner, Nanoscale, 2012, 4, 5031–5038 RSC.
- I. Shulov, S. Oncul, A. Reisch, Y. Arntz, M. Collot, Y. Mely and A. S. Klymchenko, Nanoscale, 2015, 7, 18198–18210 RSC.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1525–1568 RSC.
- X. Zhang, Z. Chen and F. Würthner, J. Am. Chem. Soc., 2007, 129, 4886–4887 CrossRef CAS PubMed; R. Abbel, R. van der Weegen, E. W. Meijer and A. P. H. J. Schenning, Chem. Commun., 2009, 1697–1699 RSC; A. Kaeser and A. P. H. J. Schenning, Adv. Mater., 2010, 22, 2985–2997 CrossRef PubMed; K. Petkau, A. Kaeser, I. Fischer, L. Brunsveld and A. P. H. J. Schenning, J. Am. Chem. Soc., 2011, 133, 17063–17071 CrossRef PubMed; J. H. Olivier, J. Widmaier and R. Ziessel, Chem. – Eur. J., 2011, 17, 11709–11714 CrossRef PubMed; X. Zhang, D. Görl and F. Würthner, Chem. Commun., 2013, 49, 8178–8180 RSC; G. Chadha, Q. Z. Yang and Y. Zhao, Chem. Commun., 2015, 51, 12939–12942 RSC.
- S. Fery-Forgues, Nanoscale, 2013, 5, 8428–8442 RSC; K. Li and B. Liu, Chem. Soc. Rev., 2014, 43, 6570–6597 RSC; O. S. Wolfbeis, Chem. Soc. Rev., 2015, 44, 4743–4768 RSC; A. Reisch and A. S. Klymchenko, Small, 2016, 12, 1968–1992 CrossRef CAS PubMed.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC; G. Battistelli, A. Cantelli, G. Guidetti, J. Manzi and M. Montalti, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2016, 8, 139–150 CrossRef CAS PubMed; Y. Niko, Y. Arntz, Y. Mely, G.-i. Konishi and A. S. Klymchenko, Chem. – Eur. J., 2014, 20, 16473–16477 CrossRef PubMed.
- F. Würthner, T. E. Kaiser and C. R. Saha-Moeller, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef PubMed.
- M. N. Berberan-Santos, P. Choppinet, A. Fedorov, L. Jullien and B. Valeur, J. Am. Chem. Soc., 1999, 121, 2526–2533 CrossRef CAS; K. A. Colby, J. J. Burdett, R. F. Frisbee, L. Zhu, R. J. Dillon and C. J. Bardeen, J. Phys. Chem. A, 2010, 114, 3471–3482 CrossRef PubMed; E. K. L. Yeow, K. P. Ghiggino, J. N. H. Reek, M. J. Crossley, A. W. Bosnian, A. P. H. J. Scheming and E. W. Meijer, J. Phys. Chem. B, 2000, 104, 2596–2606 CrossRef.
- K. Koynov and H. J. Butt, Curr. Opin. Colloid Interface Sci., 2012, 17, 377–387 CrossRef CAS.
- A. S. Klymchenko, E. Roger, N. Anton, H. Anton, I. Shulov, J. Vermot, Y. Mely and T. F. Vandamme, RSC Adv., 2012, 2, 11876–11886 RSC.
- C. Xu and W. W. Webb, J. Opt. Soc. Am. B, 1996, 13, 481–491 CrossRef CAS.
- A. Kaeser, I. Fischer, R. Abbel, P. Besenius, D. Dasgupta, M. A. J. Gillisen, G. Portale, A. L. Stevens, L. M. Herz and A. P. H. J. Schenning, ACS Nano, 2013, 7, 408–416 CrossRef CAS PubMed.
- S. W. Morton, X. Zhao, M. A. Quadir and P. T. Hammond, Biomaterials, 2014, 35, 3489–3496 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods and some experimental data. See DOI: 10.1039/c6cc03716j |
| This journal is © The Royal Society of Chemistry 2016 |