
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tetranuclear Zn/4f coordination clusters as highly efficient catalysts for Friedel–Crafts alkylation†
Kieran
Griffiths
a,
Prashant
Kumar
a,
Geoffrey R.
Akien
b,
Nicholas F.
Chilton
 c,
Alaa
Abdul-Sada
a,
Graham J.
Tizzard
d,
Simon J.
Coles
d and
George E.
Kostakis
*a
c,
Alaa
Abdul-Sada
a,
Graham J.
Tizzard
d,
Simon J.
Coles
d and
George E.
Kostakis
*a
aDepartment of Chemistry, School of Life Sciences, University of Sussex, Brighton BN1 9QJ, UK. E-mail: G.Kostakis@sussex.ac.uk
bDepartment of Chemistry, Lancaster University, Lancaster LA1 4YB, UK
cSchool of Chemistry, The University of Manchester, Manchester M13 9PL, UK
dUK National Crystallography Service, Chemistry, University of Southampton, SO171BJ, UK
First published on 18th May 2016
Abstract
A series of custom-designed, high yield, isoskeletal tetranuclear Zn/4f coordination clusters showing high efficiency as catalysts with low catalytic loadings in Friedel–Crafts alkylation are described for the first time. The possibility of altering the 4f centers in these catalysts without altering the core topology allows us to further confirm their stability via EPR and NMR, as well to gain insights into the plausible reaction mechanism, showcasing the usefulness of these bimetallic systems as catalysts.
Polynuclear coordination clusters (CCs), assembled by organic ligands, transition metal (3d) and/or lanthanide (4f) elements, represent a rapidly expanding class of materials.1–4 However, only a few and recent examples have shown the use of these materials as catalysts.5–8 Friedel–Crafts (FC) reactions9 consist of one of the most important carbon–carbon bond forming reactions in organic and medicinal chemistry. In recent years, catalysts based on zinc or lanthanide elements have been extensively employed in FC reactions and sizeable development has taken place.10,11 In particular, the indole ring system represents one of the most abundant and important heterocycles in nature exhibiting a wide range of biological activities.12 The nucleophilic addition of aldehydes or ketones to indoles, in the presence of a Lewis acid, is a facile route towards the synthesis of bisindolylmethane (BIM) derivatives with only one equivalent of water generated as a side product. BIMs have recently been shown to be useful in the treatment of fibromyalgia,13 as antibacterial agents14 and even in the prevention of cancer.14 Various catalytic systems and metal salts have been reported for the synthesis of BIMs, however these methods suffer from a number of disadvantages such as expense, toxicity of reagents, high temperatures, high catalytic loadings (stoichiometric-10%) and photosensitivity (i.e. silver salts).15–18
In 3d/4f chemistry, 4f centres may be replaced by YIII or GdIII without altering the core topology,19 therefore permitting 89Y NMR20 or EPR21 for characterization of the solution species, as well as allowing study of the catalytic reaction in situ to gain mechanistic insights. Also, the possibility of altering the 3d centres while retaining the topology22,23 may provide more insights into the reaction mechanism, due to the different coordination behaviours to substrates (heteroatoms N, O or S), anticipated to be seen when Co, Ni, Cu, or Zn is used. We recently committed to studying the catalytic properties of tetranuclear 3d/4f CCs possessing a rigid defective dicubane topology assembled solely by the Schiff base organic ligand (E)-2-(2-hydroxy-3-methoxybenzylidene-amino)phenol) H2L (Scheme S1, ESI†). We showed that a series of isoskeletal24 MII2LnIII2 (M = Co or Ni, Ln = Y, Nd, Eu, Gd, Tb, and Dy) CCs effectively catalyse a domino reaction at room temperature.8,25 These heterometallic species remain intact in organic solvents and this precise topology brings the 3d and the 4f centres very close (approximately 3.3 Å), allowing both metal centres to coordinate to the substrates and promote the coupling reaction in high yield, highlighting the catalytic utility of this tetranuclear bimetallic motif.
Motivated by our previous results, and having in mind that Ln(OTf)326 and Zn(ClO)4/Schiff base27 compounds have been employed as catalysts in the reaction of indole derivatives with aldehydes and ketones with a loading of 10 mol%, we decided to synthesize and characterize a series of isoskeletal ZnII2LnIII2 CCs and study, for the first time, their application in FC reactions.
The combination under aerobic conditions of H2L with freshly prepared Ln(NO3)3·xH2O and Zn(NO3)2·6H2O in the presence of Et3N in EtOH afforded, in very good yields, precipitates subsequently crystallized by vapour diffusion of Et2O in N,N′-DMF solutions, affording isoskeletal air-stable tetranuclear defective dicubane compounds with the general formula [ZnII2LnIII2L4(NO3)2(DMF)2] where Ln is Y (1Y), Sm (1Sm), Eu (1Eu) Gd (1Gd), Dy (1Dy), Tb (1Tb) and Yb (1Yb) (Fig. 1). These compounds were characterized in full by X-ray crystallography (Tables S1 and S2, ESI†), IR, ESI-MS, TGA and elemental analysis (see the ESI†).
 | ||
| Fig. 1 Molecular structure of 1Ln. Colour codes: ZnII: grey; LnIII: light blue; O: red; N: blue; C: yellow. Hydrogen atoms are omitted for clarity. | ||
The ZnII ion adopts a slightly distorted octahedral geometry with an O5N donor set, while the Ln ion has a very distorted square antiprismatic geometry, by virtue of chelating nitrate anions, with an O7N donor set. Five out of six (Zn) and six out of eight (Ln) coordination sites are occupied by donors from the organic ligands. The remaining coordination sites are occupied by one DMF molecule (Zn) and one nitrate anion (Ln, chelated mode). For 1Y, the Y⋯Zn distances are 3.34882(7) and 3.5272(8) Å and the Zn⋯Zn distance is 3.2949(8) Å; the other 1Ln species are similar.
The identity of 1Ln in solution was confirmed by electrospray ionization mass spectrometry (ESI-MS) studies; in all cases, we observed two peaks in the MS (positive-ion mode) at m/z which corresponds perfectly to the monocationic [ZnII2LnIII2L4(NO3)]+ and the dicationic [ZnII2LnIII2L4]2+ fragments, respectively (see Fig. S2–S19, ESI†).
For further confirmation of the solution stability of 1Ln, Q-band EPR studies of 1Gd in both solid and solution (80% DMF and 20% Et2O) phases were performed, Fig. 2. Simulations with PHI28 confirm that the spectra owe to S = 7/2 GdIII ions with rhombic zero-field splitting (ZFS) (see Fig. S1, ESI†). The highly sensitive, finger-print-like ZFS of the S = 7/2 state directly indicates that the coordination environment of the GdIII ion remains unchanged in solution.
 | ||
| Fig. 2 Experimental EPR spectra at the Q-band (34.0865 GHz) of 1Gd, recorded at 7 K in the solid (green line) and solution phase (red line). | ||
For benchmarking studies, indoles and benzaldehyde were selected as reactants using 1Dy as the catalyst at 10% loading, and the reaction parameters were subsequently optimized.‡ The first set of reactions was performed in order to identify the ideal solvent system; after screening several solvents (Table S3, ESI†), we identified that the ethanol/water (2/1) solvent system (Table S3, ESI,† entry 9) provided the best catalytic yield. The use of DMF, acetonitrile or EtOH as solvent (Table S3, ESI,† entries 4–6) resulted in the same product but in lower yields, indicating that the presence of H2O is crucial to obtain high yields. No conversion to products was observed in low polarity solvents in which 1Dy is insoluble (Table S3, ESI,† entries 1–3), suggesting that the catalytic action is homogeneous. We then performed a series of reactions at a 2.5% catalyst loading and varying the H2O/EtOH ratio (Table S4, ESI†) and quantitative yields (entries 5–6) were obtained in the 3/2 and 1/1 EtOH/H2O ratio. After selecting the optimum solvent system, we conducted reactions without the 1Dy catalyst (Table 1, entry 1) and no product was obtained. Next, due to the heteronuclear (Zn/Ln) character of our molecules, lanthanide and zinc triflates were utilized at 10 mol% loading (Table 1, entries 2–5), in order to identify their influence on the bisindolylalkane product.
| Entrya | Catalyst | Loadingb/mol% | Time/h | Yieldc/% |
|---|---|---|---|---|
a Reaction conditions: indole, 1 mmol; benzaldehyde, 0.5 mmol; catalyst; 10 mL EtOH/H2O (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1); room temperature.
b Catalyst loading calculated per equivalent of LnIII.
c Determined by 1H NMR spectroscopy. :1); room temperature.
b Catalyst loading calculated per equivalent of LnIII.
c Determined by 1H NMR spectroscopy.
|
||||
| 1 | None | N/A | 12 | 0 |
| 2 | Dy(OTf)3 | 10 | 12 | 95 |
| 3 | Y(OTf)3 | 10 | 12 | Quantitative |
| 4 | Y(OTf)3 | 2.5 | 12 | 55 |
| 5 | Zn(OTf)3 | 10 | 12 | 18 |
| 6 | 1Dy | 10 | 2 | Quantitative |
| 7 | 1Dy | 5 | 2 | 96 |
| 8 | 1Dy | 2.5 | 12 | 92 |
| 9 | 1Dy | 1 | 12 | 87 |
| 10 | 1Y | 2.5 | 12 | Quantitative |
| 11 | 1Y | 1 | 12 | 96 |
| 12 | 1Sm | 2.5 | 12 | 87 |
| 13 | 1Eu | 2.5 | 12 | 53 |
| 14 | 1Tb | 2.5 | 12 | 80 |
| 15 | 1Gd | 2.5 | 12 | 75 |
| 16 | 1Yb | 2.5 | 12 | 94 |
Yttrium triflate (Table 1, entry 3) was found to give quantitative yields and dysprosium triflate (Table 1, entry 2) produced only slightly lower yields, while zinc triflate (Table 1, entry 5) showed poor catalytic performance. Compound 1Dy afforded almost quantitative yields of the desired product after 2 h at 10 mol% loading. By use of 1Sm, 1Eu, 1Tb, 1Gd, and 1Yb comparable performance to 1Dy was obtained, however the use of 1Y at 2.5 mol% loading afforded the desired material in quantitative yield (Table 1, entry 10). Promisingly, the catalyst loading for 1Y could be decreased to 1 mol% with only a slight decrease in the yields (Table 1, entry 11), being far lower than other reported systems.16,17,27 Importantly, reducing the catalyst loading for yttrium triflate to 2.5 mol% (Table 1, entry 4) resulted in a severe decrease in yield, compared to the ability of 1Dy and 1Y to maintain high yields at only a few mol%.
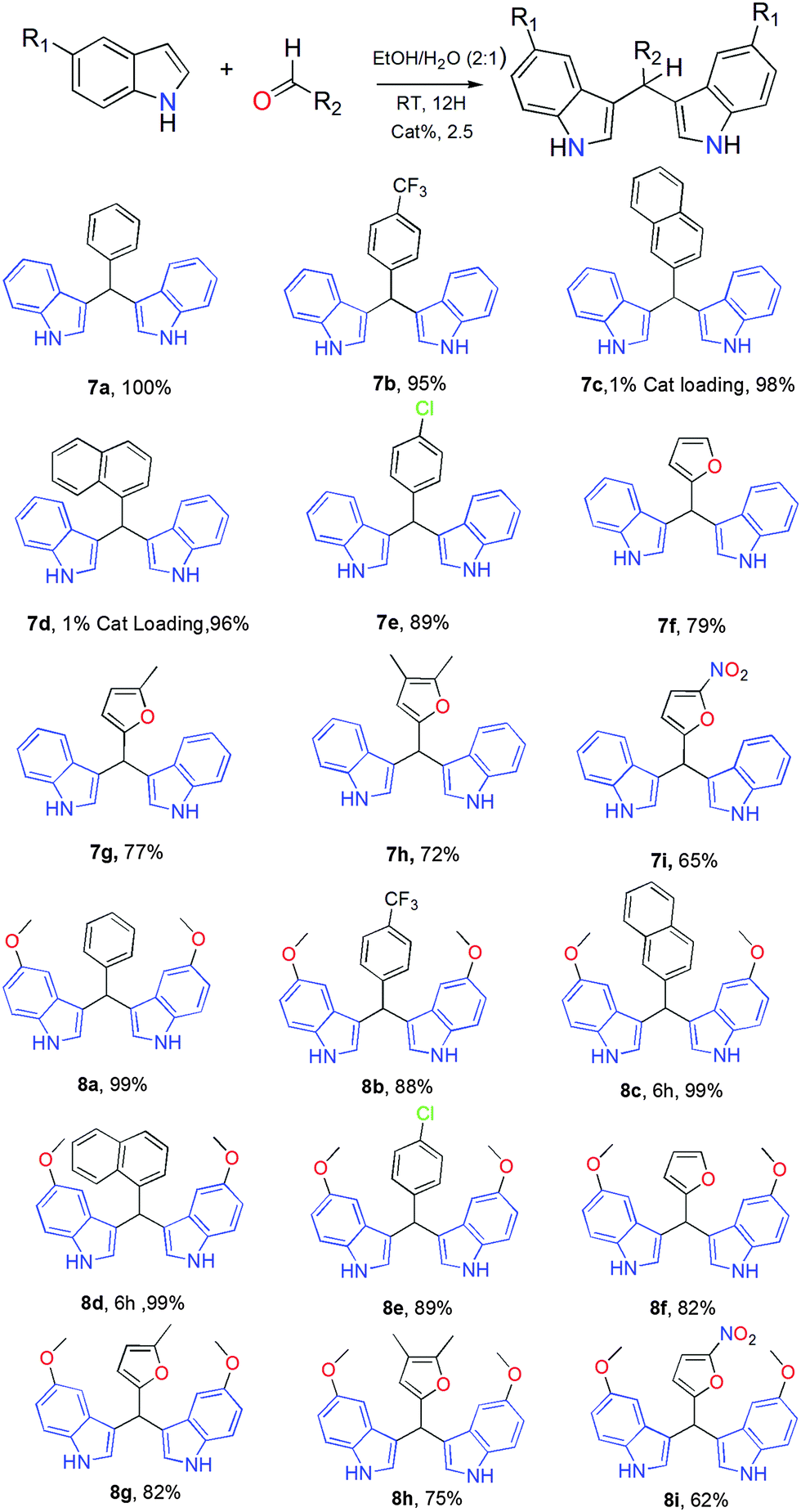
We then explored the scope of the reaction by employing a variety of aldehydes and substituted indoles (Table 2). The reaction proceeds smoothly with very good to excellent yields. Products 7a, 8c and 8f were characterized via single crystal X-ray diffraction (Fig. S20, ESI†). The next step was to involve ketones in place of aldehydes. Long reaction times even with a slight increase of the temperature (50 °C) did not yield any product with both aliphatic (acetone, cyclohexanone) and aromatic ketones (acetophenone) and no unexpected side products were observed. This opposite behaviour recalls the selective reduction of ketones in the presence of aldehydes (Luche reaction).29 In this reaction, aldehydes bond to Ln centres via hemiketal form, whilst ketone remains unprotected, and is thus reduced by NaBH4. Efforts to monitor the formation of the hemiketal form by NMR were not successful. To gain information on the reaction mechanism we performed UV-Vis binding studies of 2-naphthaldehyde with Zn(OTf)2, Dy(OTf)3 and 1Dy (Fig. S21, ESI†). 1Dy is found to have almost double the rate of 2-naphthaldehyde quenching compared to Dy(OTf)3, and Zn(OTf)2 is significantly slower than both. This observation indicates that 2-naphthaldehyde prefers coordination to Dy over Zn.
|
a Reaction conditions: aldehyde, 0.5 mmol; substituted indole, 1 mmol; EtOH/H2O, (2:1) 10 mL; room temperature; 12 h. 2.5% loading of 1Y.
|
|---|
|
To gain further information on the plausible mechanism and to identify the limitations of the present catalysts, we decided to perform the following set of reactions. First, we employed an aliphatic aldehyde (cyclohexanecarbaldehyde) instead of an aromatic aldehyde in reaction with indoles, which gave very low yields of the expected product 9 (Scheme S1, ESI†) after 72 hours. Second, we examined substituted indoles such as 2-methyl-indole, 2-(trifluoromethyl)-indole, 3-methyl indole, indole-3-acetic acid and N-methyl indole in reaction with benzaldehyde. The use of 2-methyl-indole gave the expected product 10 (Scheme S1, ESI†) in quantitative yield in only 2 hours, indicating that substitution in position 2 promotes the reaction. Compound 10 was characterized via single crystal X-ray diffraction (Fig. S22, ESI†). However, the use of the electron withdrawing group –CF3 in place of –CH3 [reaction with 2-(trifluoromethyl)-indole] gave a yield of only 16%, product 11, indicating a substantial influence on the catalytic activity. The use of indole-3-acetic acid did not yield any product. A logical explanation for deterioration of catalytic activity is that the carboxylic group of indole-3-acetic acid is in competition with benzaldehyde to coordinate to the Ln centre, leading to a poisoning of the catalyst, however the use of 3-methyl-indole did not result in the formation of BIM. The latter indicates that if the most active site (C-3) of the indole group is blocked the reaction does not proceed. Finally, the reaction of N-methyl-indole with benzaldehyde did not yield any product, showcasing that coordination of the nitrogen atom is crucial.
Despite the diamagnetic nature of 1Y, characterizing it in the solution state by NMR proved to be difficult due to its asymmetry, dynamic behaviour, and its relatively low solubility. However, the peak areas for the imine, aromatic, and methoxy protons were all consistent with the structure as determined by X-ray crystallography, Fig. 1. Gradually warming to 75 °C in 10 °C steps caused gradual broadening of the peaks, which then recoalesced upon cooling back to room temperature, without any apparent decomposition of the complex. Using multiple solvent suppression, it was possible to monitor the smooth conversion of benzaldehyde and indole to the product in protonated 2:1 EtOH/H2O. With Y(OTf)3, the concentration of intermediates appeared to be very low, but with prolonged acquisition it was possible to identify a minor species using 15N-HMBC. Its chemical shift of 113 ppm is distinctly different from indole (130 ppm) and the product (125 ppm), but the proton chemical shifts of 7.33 (127.9 ppm in 13C-HSQC) and 4.99 ppm (too close to the solvent peak to identify 13C-correlations) suggest that it may be the benzaldehyde-indole hemiaminal. In the absence of a catalyst, the product forms very slowly (ca. 10% in 1 month), but under the same spectroscopic conditions it was not possible to detect the same correlations. This raises the prospect that it may well be a key intermediate, but more favourable conditions may be required to unambiguously identify it.
Given knowledge of the structure of the catalyst and the data provided above, we propose the following mechanism for the FC reaction (Scheme 1). The first step of the reaction involves the coordination of the Nindole atom to the ZnII and the Ocarbonyl atom of the aldehyde to the Ln part of the catalyst (Step 1). In the presence of H2O the aldehyde may coordinate to the Ln centre via its hydrated form. Then, deprotonation of the coordinated indole leads to the formation of a negative centre at C-3 of the indole moiety (Step 2). The two organic moieties are very close [Y⋯Zn distances are 3.34882(7) and 3.5272(8) Å] to favour the formation of benzaldehyde-indole hemiaminal, as suggested by the NMR studies (Steps 3 and 4) followed by alkylation of one more indole moiety (Step 5). Finally, the catalytic cycle ends with a proton exchange with an additional indole moiety (Step 6) and release of the bis-adduct product and water and reform of the active catalyst species (Step 7).
 | ||
| Scheme 1 A plausible mechanism for the FC alkylation. | ||
Overall, the results presented herein confirm for the first time that these tetranuclear CCs may indeed be useful as FC catalysts, with low catalyst loadings. Our philosophy to alter the 4f elements in these solution-stable bimetallic 3d/4f species allow us to confirm their stability via EPR and gain useful insights into the plausible reaction mechanism via NMR. Moreover, the possibility of tuning the organic periphery of these catalysts,8,25 achieving immobilization, showcases that the present catalysts are highly desirable and appropriate vehicles to open new avenues in the fields of 3d/4f chemistry and catalysis. Further work is necessary to expand the scope of the present Zn/Ln CCs to other FC alkylation reactions, and this will be the focus of our future activities.
We thank the EPSRC (UK) for funding (grant number EP/M023834/1), the University of Sussex, for offering a PhD position to K. G., the EPSRC UK National Crystallography Service at the University of Southampton30 for the collection of the crystallographic data for compounds 7a, 8c, 8f and 10, and the EPSRC UK National Electron Paramagnetic Resonance Service at The University of Manchester.
Notes and references
- J. Jankolovits, C. M. Andolina, J. W. Kampf, K. N. Raymond and V. L. Pecoraro, Angew. Chem., Int. Ed., 2011, 50, 9660–9664 CrossRef CAS PubMed.
- S. Ahmed, M. A. Mansoor, M. Mazhar, T. Söhnel, H. Khaledi, W. J. Basirun, Z. Arifin, S. Abubakar and B. Muhammad, Dalton Trans., 2014, 43, 8523–8529 RSC.
- X.-J. Kong, L.-S. Long, R.-B. Huang, L.-S. Zheng, T. D. Harris and Z. Zheng, Chem. Commun., 2009, 43544356 Search PubMed.
- K. Liu, W. Shi and P. Cheng, Coord. Chem. Rev., 2015, 289–290, 74–122 CrossRef CAS.
- F. Evangelisti, R. More, F. Hodel, S. Luber and G. R. Patzke, J. Am. Chem. Soc., 2015, 137, 11076–11084 CrossRef CAS PubMed.
- S. Handa, V. Gnanadesikan, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2007, 129, 4900–4901 CrossRef CAS PubMed.
- G. Maayan and G. Christou, Inorg. Chem., 2011, 50, 7015–7021 CrossRef CAS PubMed.
- K. Griffiths, C. W. D. Gallop, A. Abdul-Sada, A. Vargas, O. Navarro and G. E. Kostakis, Chem. – Eur. J., 2015, 21, 6358–6361 CrossRef CAS PubMed.
- C. Friedel and J. M. Crafts, Compt. Rend., 1877, 1392–1395 Search PubMed.
- Y. Hui, L. Lin, X. Liu and X. Feng, Zinc Catalysis: Applications in Organic Synthesis, WILEY-VCH Verlag, 2015 Search PubMed.
- G. K. Veits and J. Read de Alaniz, Tetrahedron, 2012, 68, 2015–2026 CrossRef CAS.
- M. Inman and C. J. Moody, Chem. Sci., 2013, 4, 29–41 RSC.
- S. Sarva, J. S. Harinath, S. P. Sthanikam, S. Ethiraj, M. Vaithiyalingam and S. R. Cirandur, Chin. Chem. Lett., 2016, 27, 16–20 CrossRef CAS.
- V. Jamsheena, G. Shilpa, J. Saranya, N. A. Harry, R. S. Lankalapalli and S. Priya, Chem.-Biol. Interact., 2016, 247, 11–21 CrossRef CAS PubMed.
- C. Karami, H. Ahmadian, M. Nouri, F. Jamshidi, H. Mohammadi, K. Ghodrati, A. Farrokhi and Z. Hamidi, Catal. Commun., 2012, 27, 92–96 CrossRef CAS.
- S. Khaksar, M. Tajbakhsh and M. Gholami, C. R. Chim., 2014, 17, 30–34 CrossRef CAS.
- M. A. Zolfigol, P. Salehi, M. Shiri and Z. Tanbakouchian, Catal. Commun., 2007, 8, 173–178 CrossRef CAS.
- J. Beltrá, M. C. Gimeno and R. P. Herrera, Beilstein J. Org. Chem., 2014, 10, 2206–2214 CrossRef PubMed.
- V. Mereacre, A. M. Ako, R. Clérac, W. Wernsdorfer, I. J. Hewitt, C. E. Anson and A. K. Powell, Chem. – Eur. J., 2008, 14, 3577–3584 CrossRef CAS PubMed.
- M. W. Löble, M. Casimiro, D. T. Thielemann, P. Oña-Burgos, I. Fernández, P. W. Roesky and F. Breher, Chem. – Eur. J., 2012, 18, 5325–5334 CrossRef PubMed.
- S. Rast, A. Borel, L. Helm, E. Belorizky, P. H. Fries and A. E. Merbach, J. Am. Chem. Soc., 2001, 123, 2637–2644 CrossRef CAS PubMed.
- A. M. Ako, V. Mereacre, R. Clérac, I. J. Hewitt, Y. Lan, C. E. Anson and A. K. Powell, Dalton Trans., 2007, 5245–5247 RSC.
- A. M. Ako, V. Mereacre, R. Clérac, I. J. Hewitt, Y. Lan, G. Buth, C. E. Anson and A. K. Powell, Inorg. Chem., 2009, 48, 6713–6723 CrossRef CAS PubMed.
- K. Griffiths, V. N. Dokorou, J. Spencer, A. Abdul-Sada, A. Vargas and G. E. Kostakis, CrystEngComm, 2016, 18, 704–713 RSC.
- K. Griffiths, P. Kumar, J. D. Mattock, A. Abdul-Sada, M. B. Pitak, S. J. Coles, O. Navarro, A. Vargas and G. E. Kostakis, Inorg. Chem. Search PubMed , under revision.
- D. Chen, L. Yu and P. G. Wang, Tetrahedron Lett., 1996, 37, 4467–4470 CrossRef CAS.
- Y. H. Hui, Y. C. Chen, H. W. Gong and Z. F. Xie, Chin. Chem. Lett., 2014, 25, 163–165 CrossRef CAS.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 1164–1175 CrossRef CAS PubMed.
- A. L. Gemal and J. L. Luche, J. Am. Chem. Soc., 1981, 103, 5454–5459 CrossRef CAS.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683–689 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1452416–1452423. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc03608b |
| ‡ Catalytic protocol. To an EtOH/H2O (2:1 in 10 mL) mixed solution, aldehyde (0.5 mmol) and indole (1 mmol) were added followed by the catalyst (2.5% with respect to aldehyde). The resultant solution was stirred for 12 h, during which time the product had precipitated. The cloudy solution was filtered and the precipitate was washed with hexane (3 × 10 mL) and water (3 × 10 mL). For products which did not precipitate, the clear solution was concentrated under reduced pressure and extracted in ethyl acetate (30 mL) from water (20 mL). The ethyl acetate layer was dried with MgSO4 and concentrated under reduced pressure. The resulting oil was washed with hexane (60 mL), resulting in a powdered solid which was further washed with water (3 × 10 mL). |
| This journal is © The Royal Society of Chemistry 2016 |