
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cyclic dinucleotide (c-di-GMP, c-di-AMP, and cGAMP) signalings have come of age to be inhibited by small molecules
Clement
Opoku-Temeng
ab,
Jie
Zhou
a,
Yue
Zheng
ab,
Jianmei
Su
b and
Herman O.
Sintim
*a
aDepartment of Chemistry, Center for Drug Discovery, Purdue University, West Lafayette, IN 47907, USA. E-mail: hsintim@purdue.edu; Tel: +1 (765) 494-5200
bGraduate Program in Biochemistry, University of Maryland, College Park, Maryland 20742, USA
First published on 8th June 2016
Abstract
Bacteria utilize nucleotide-based second messengers to regulate a myriad of physiological processes. Cyclic dinucleotides have emerged as central regulators of bacterial physiology, controlling processes ranging from cell wall homeostasis to virulence production, and so far over thousands of manuscripts have provided biological insights into c-di-NMP signaling. The development of small molecule inhibitors of c-di-NMP signaling has significantly lagged behind. Recent developments in assays that allow for high-throughput screening of inhibitors suggest that the time is right for a concerted effort to identify inhibitors of these fascinating second messengers. Herein, we review c-di-NMP signaling and small molecules that have been developed to inhibit cyclic dinucleotide-related enzymes.
Introduction
The ability of cells to sense and respond to environmental cues is critical for survival. Signals such as nutrition, stress, changes in temperature, osmolarity and pH must be precisely sensed and adequately responded to. Second messengers are crucial in the transduction of signals and nucleotide-based second messengers have over the years been demonstrated to be central to this process (Fig. 1).1 Cyclic mononucleotides, such as cAMP and cGMP (Fig. 2), are key second messengers in both bacteria and higher organisms. For example, cAMP controls carbon metabolism, motility and virulence.2 cGMP has also been shown to control a variety of key biological processes, such as smooth muscle relaxation and regulation of the intestinal electrolyte and fluid homeostasis in mammals.3 In bacteria, cGMP has been shown to regulate the development of Rhodospirillum centenum, an alphaproteobacterium.4 The alarmones, guanosine tetraphosphate and guanosine pentaphosphate ((p)ppGpp), have been shown to regulate stringent response in bacteria.5,6 Nicotinic acid adenine dinucleotide phosphate (NAADP), adenosine diphosphoribose (ADPR), and cyclic ADP-ribose (cADPR) function as Ca2+ mobilizers that affect various cellular processes.7 The cADPR analog, cyclic inosine diphosphoribose ether (cIDPRE), has also been shown to cause Ca2+ release in human Jurkat T-lymphocytes.8 Dinucleotides, such as NAD and NADP, are also important signaling molecules and are involved in T-cell calcium signaling.7 Diadenosine polyphosphates (Ap2A, Ap3A, Ap4A, Ap5A and Ap6A) are also ubiquitous second messengers with physiological roles ranging from serving as alarmones to being vasoactive molecules.9–11Fig. 2 shows the structures of some of the well-established nucleotide second messengers.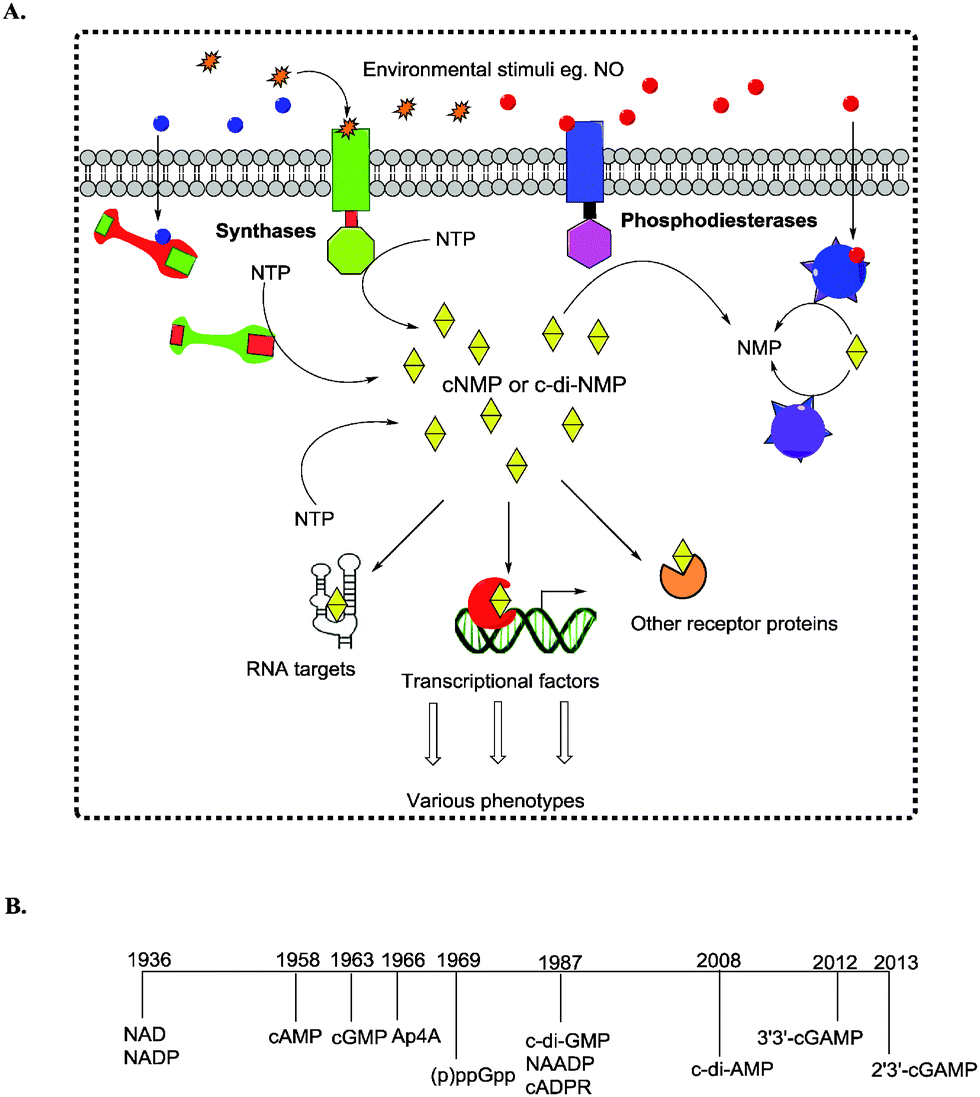 | ||
| Fig. 1 (A) Signal transduction from a primary signal (first messengers) to second messengers that relay and amplify information to macromolecule regulation that affects the phenotype. (B) Timeline of the discovery of nucleotide-based second messengers. | ||
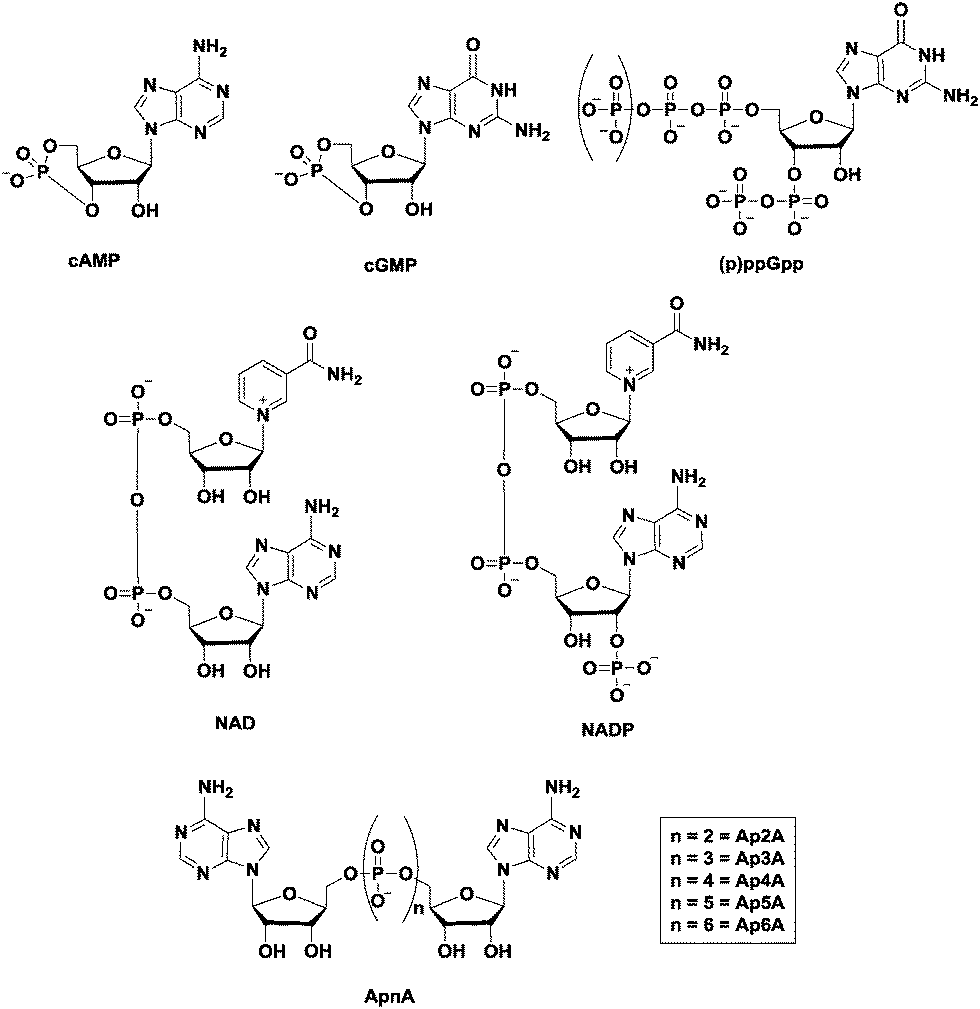 | ||
| Fig. 2 Structures of some of the well-established nucleotide second messengers. | ||
Due to the importance of nucleotide second messengers in biology, massive efforts have been focused on the development of small molecules that inhibit the signaling of these second messengers. The majority of inhibitor developments against nucleotide second messengers had focused on cAMP/cGMP and several small molecules that inhibit the phosphodiesterases of cyclic nucleotide monophosphate are now used in the clinic (see Fig. 3 for examples).
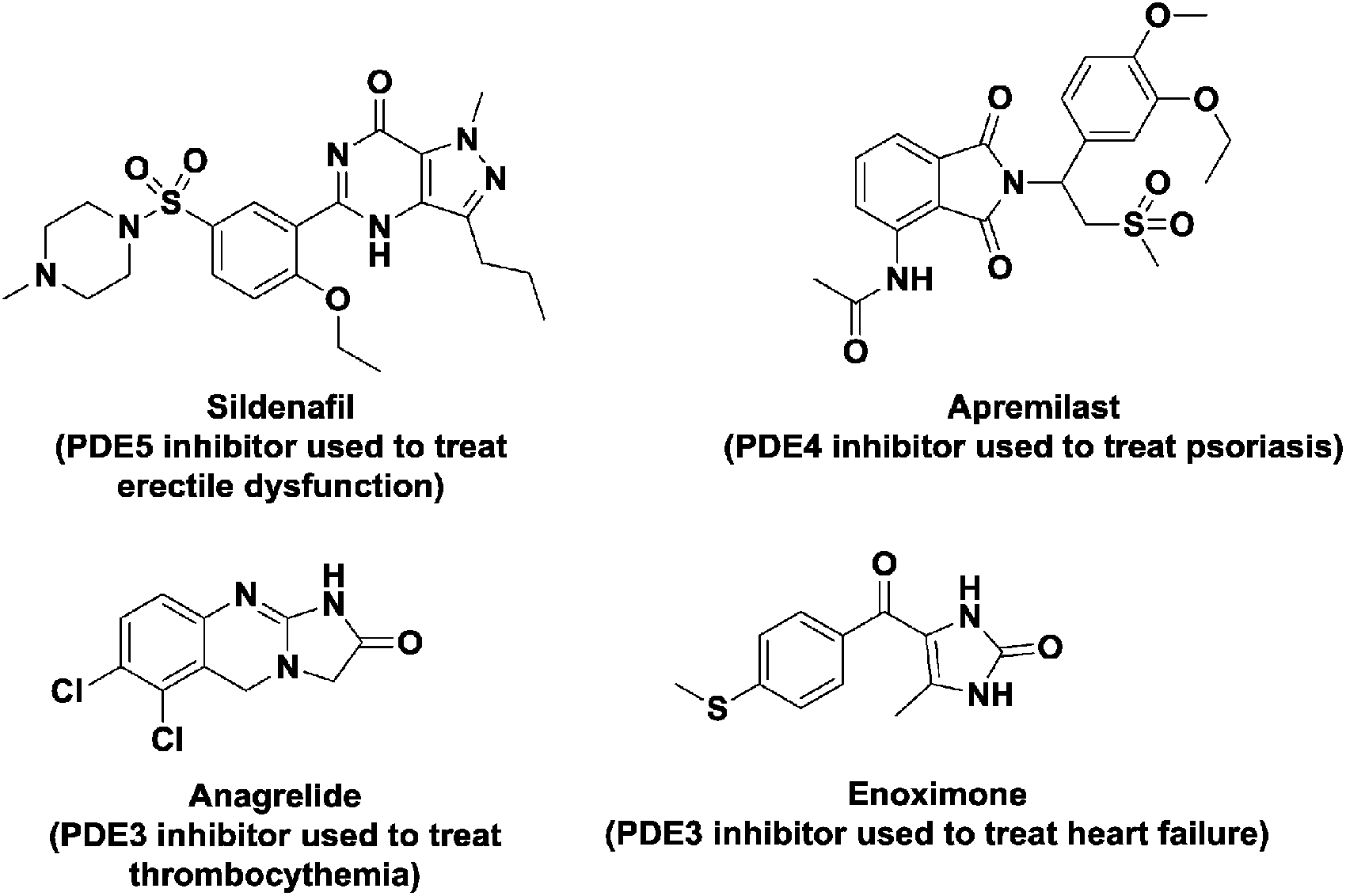 | ||
| Fig. 3 Structures of some PDE inhibitors and their clinical uses. | ||
Cyclic dinucleotides (see Fig. 4 for structures) have now emerged as important second messengers and the dazzling arrays of processes that these molecules appear to regulate suggest that these dinucleotides will also become as important as cyclic nucleotide monophosphates.1,12–19 The first cyclic dinucleotide, c-di-GMP, was the first to be discovered by Benziman in 1987 and has since been established as the master regulator of the bacterial lifestyle in mostly Gram-negative and a handful of Gram-positive bacteria.1,20 c-di-AMP was also later identified to be a crucial second messenger in Gram-positive bacteria and mycobacteria.1,21 Subsequently, hybrid cyclic dinucleotides cGAMP (2′3′ and 3′3′)22,23 were also identified to regulate various processes.24,25 Efforts towards understanding the molecular mechanisms underlying the activities of cyclic dinucleotides have uncovered a myriad of cyclic dinucleotide synthases, phosphodiesterases, effector proteins and RNA, all of which work in a concerted manner to execute a specific action in response to the cyclic dinucleotide second messengers.13,21,26
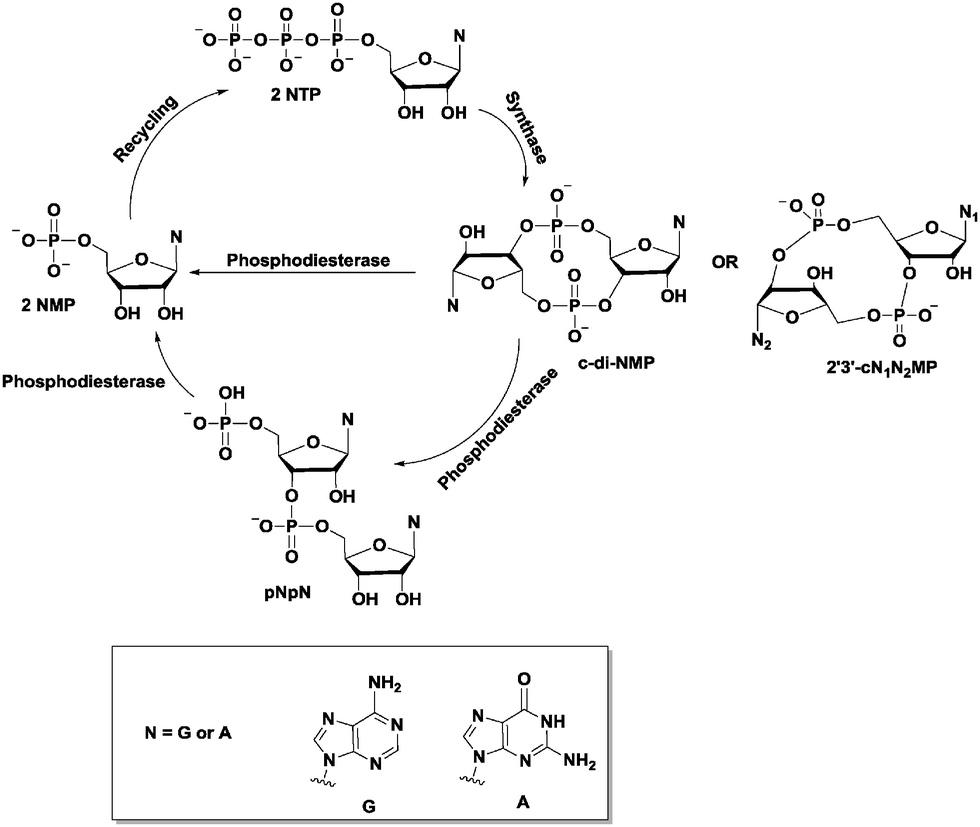 | ||
| Fig. 4 Schematic of the general synthesis and degradation of cyclic dinucleotide second messengers. | ||
Due to the seemingly indispensable roles of cyclic dinucleotides in both bacteria and higher organisms (in this case as immune response regulators), research has also been directed towards ways of interrupting cyclic dinucleotide signaling. Consequently, various inhibitors have been discovered/developed against cyclic dinucleotide signaling. The field of cyclic dinucleotide inhibitor discovery is now in its infancy with only a handful of groups pioneering inhibitor development but it is projected that there will be an explosion of research activities towards the development of small molecule regulators of cyclic dinucleotide signaling as it becomes increasingly clear that these second messengers are critical for various biological processes and diseased states. In this review, we discuss some of the fundamentals of cyclic dinucleotide signaling with emphasis on inhibitors developed against the cyclic dinucleotides.
c-di-GMP
Bis-(3′–5′) cyclic diguanylic acid (c-di-GMP) was originally discovered by Benziman and colleagues in 1987 as an activator of cellulose synthase in a Gram-negative bacterium, Gluconoacetobacter xylinus (formerly named Acetobacter xylinum).20 Although it was discovered almost 30 years ago, it is only in the last decade that the many cellular processes that are regulated by c-di-GMP have been elucidated. The major function of c-di-GMP is the regulation of motility-to-sessility transition.1 It is also a key player in the cell cycle and virulence factor production.1 So far, c-di-GMP has been identified in a wide variety of Gram-negative bacteria, including Pseudomonas aeruginosa, Caulobacter crescentus, Escherichia coli, Salmonella typhimurium and Vibrio cholerae, and a few Gram-positive bacteria, such as Bacillus subtilis and Listeria monocytogenes.1c-di-GMP metabolism enzymes
Since c-di-GMP plays important roles in the cell, there is interest in modulating the intracellular concentrations of the dinucleotide with small molecules. The intracellular concentration of c-di-GMP is fine-tuned by its metabolism enzymes, diguanylate cyclase (DGC) and phosphodiesterase (PDE) (Fig. 4–7). DGCs cyclize two molecules of GTP to c-di-GMP and release two molecules of pyrophosphates. DGC was first purified and tested in vitro by the Benziman group.20 In 1995, Newton and colleagues characterized the gene of a DGC PleD in Caulobacter crescentus and identified a novel C-terminal GGDEF (Gly-Gly-Asp-Glu-Phe) domain.27 The conserved GGDEF or GGEEF (Gly-Gly-Glu-Glu-Phe) motifs were confirmed to be commonly shared by most DGCs.28–30 Some well characterized examples are WspR29 from P. aeruginosa (PDB 3I5A) and DosC31 (also known as YddV) from E. coli (PDB 4ZVE). Some non-canonical domains also display DGC functions, such as the AGDEF domain in V. cholerae VCA096532 and the SGDEF domain in Pectobacterium atrosepticum ECA3270.33 Besides the active site (A-site), some DGCs also contain an inhibitory site (I-site). When c-di-GMP binds to the I-site of the DGC, characterized by the RxxD (x refers to any amino acid) sequence, c-di-GMP synthesis is allosterically inhibited.34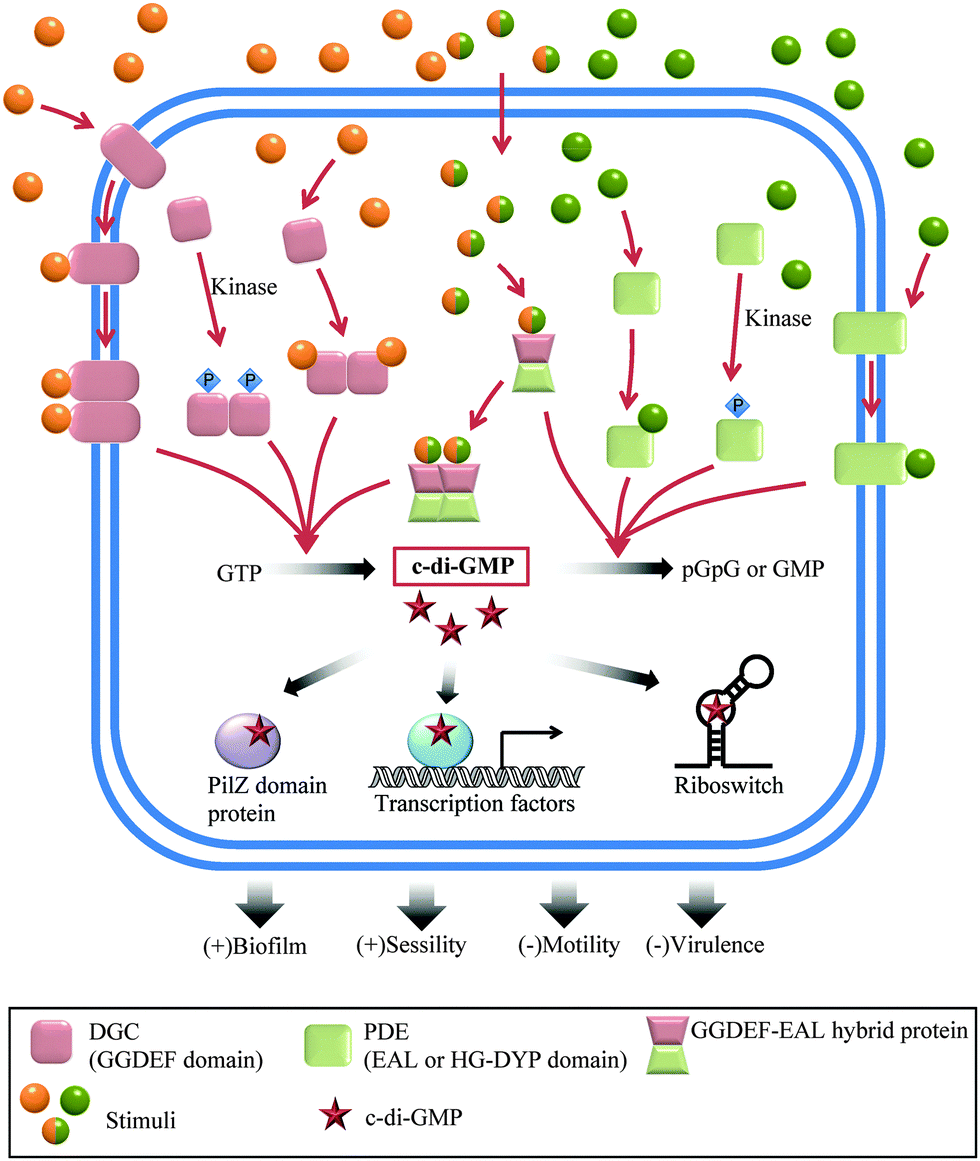 | ||
| Fig. 5 Overview of c-di-GMP regulation in bacteria. | ||
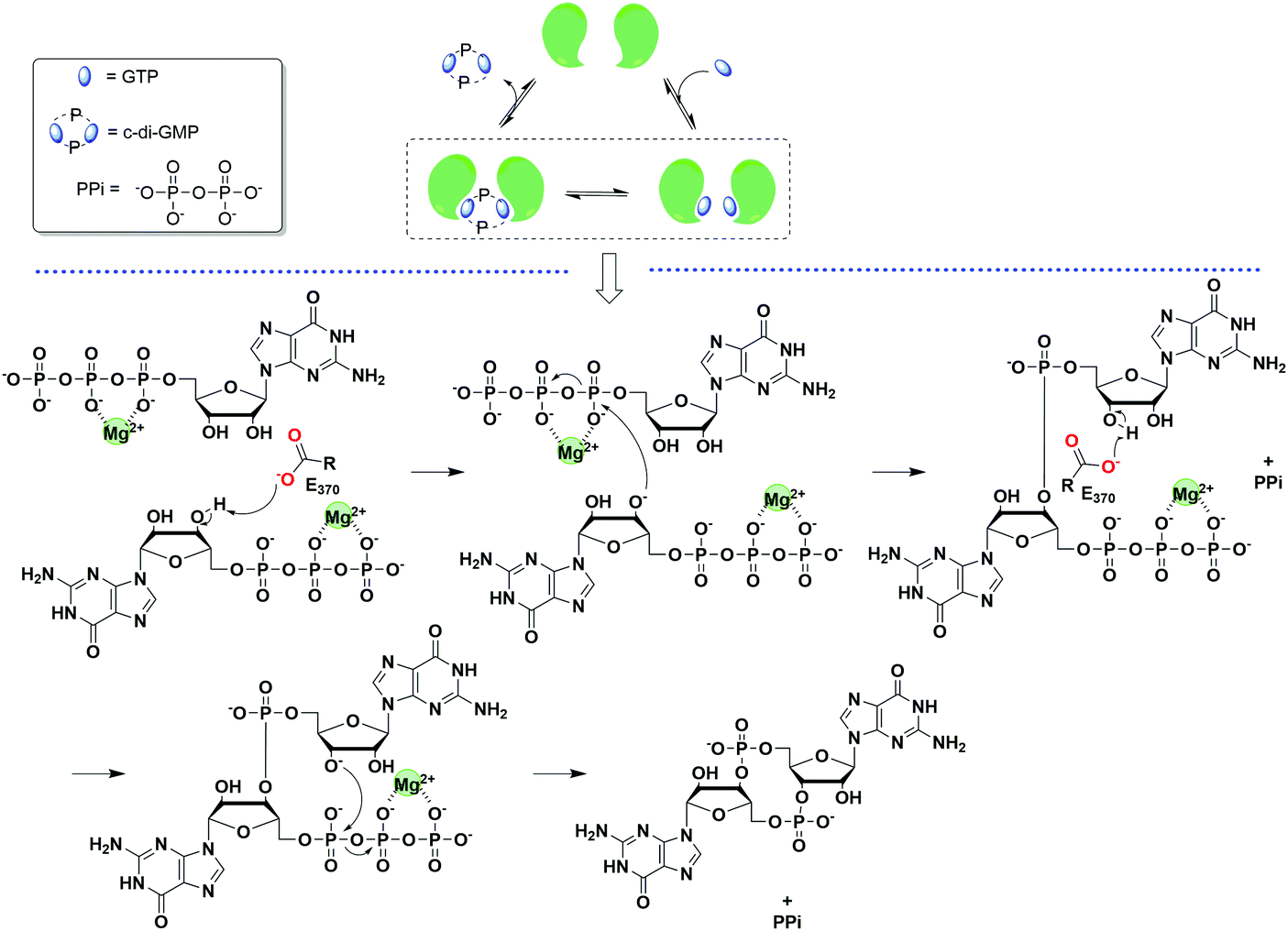 | ||
| Fig. 6 General scheme of the mechanism of c-di-GMP synthesis. The example was taken from PleD.35 (PDB 1W25). | ||
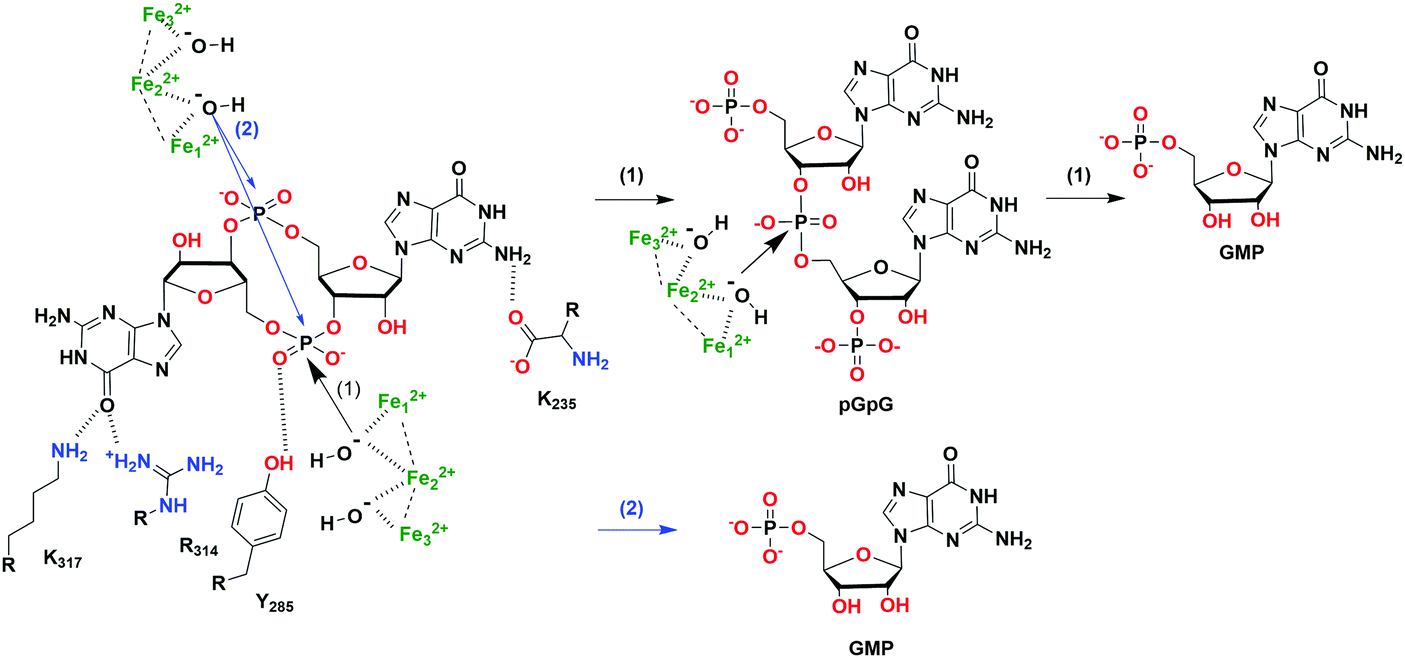 | ||
| Fig. 7 The hydrolysis mechanism of c-di-GMP by the HD-GYP domain protein from Persephonella marina (PmGH).46 (PDB 4MDZ, PmGH in complex with c-di-GMP). Scheme (1) (black arrows) indicates a two-step hydrolysis of c-di-GMP, an attack by one activated water molecule on one phosphodiester linkage forms pGpG which is further degraded into GMP; scheme (2) (blue arrows) indicates a one-step hydrolysis of c-di-GMP, an attack by two activated water molecules on both phosphodiester linkages directly forms GMP. | ||
The hydrolysis of c-di-GMP is conducted by two types of phosphodiesterases (PDEs): EAL (Glu-Ala-Leu) domain PDE and HD-GYP (His-Asp and Gly-Tyr-Pro) domain PDE (Fig. 2 and 5). The major product of EAL domain PDE is 5′-phosphoguanylyl-guanosine (pGpG) and the degradation of pGpG to guanosine monophosphate (GMP) is slow. For example, RocR (PDB 3SY8)36 from P. aeruginosa cleaves c-di-GMP into pGpG in the presence of Mg2+ or Mn2+ and this reaction is inhibited by Ca2+ and Zn2+ ions.37 Recently it was disclosed that in bacteria, such as P. aeruginosa, which harbour PDEs that mainly cleave c-di-GMP into pGpG, a second PDE (an oligoribonuclease) completes the cleavage of pGpG into GMP.38,39 The role of oligoribonuclease in cyclic dinucleotide metabolism is expected because these enzymes (found in several bacteria) have long been known to cleave short oligonucleotides into NMPs.40–42 Galperin and colleagues predicted the functions of HD-GYP in c-di-GMP hydrolysis by bioinformatic studies43 and the biochemical evidence for this hypothesis was shown by Dow and colleagues.44 The HD-GYP domain PDE RpfG from Xanthomonas campestris pv. campestris directly cleaves c-di-GMP into GMP.44 There are a few crystal structures of HD-GYP PDEs reported. Bd1817 from Bdellovibrio bacteriovorus (PDE 3TM8), which lacks the tyrosine in the GYP active site, presents a bi-iron center.45 However, the structure of another HD-GYP protein PmGH from Persephonella marina shows a different trinuclear iron active site (PDB 4ME4).46 In 2015, the structure of PA4781 from Pseudomonas aeruginosa was reported (PDB 4R8Z) and the bimetallic active site binds to Mn2+, Ni2+ and some other transition metals with similar affinities.47
c-di-GMP receptors
Different types of c-di-GMP binding proteins have been identified, augmenting the important roles played by c-di-GMP in the bacterial lifestyle (planktonic or sessile).13 When the intracellular concentration of c-di-GMP is high, it induces extracellular polymeric substance (EPS) production and promotes biofilm formation.48 When c-di-GMP concentration is low, flagella biosynthesis is upregulated and bacterial motility significantly increases.49 c-di-GMP drives the C. crescentus cell cycle by inducing the dephosphorylation or degradation of a replication initiation inhibitor CtrA through the effector proteins CckA and PopA respectively.50,51 c-di-GMP also represses acute virulence gene transcription.52 For some intracellular pathogens, c-di-GMP has been shown to make bacterial cell surface proteins resistant to proteolytic cleavage, enhancing bacterial invasion.53 There are three major types of c-di-GMP binding proteins: (1) the PilZ domain c-di-GMP receptor; (2) DGC I-sites and inactive EAL and HD-GYP domains; and (3) other types of c-di-GMP receptors.13The first type of c-di-GMP binding domain PilZ (Pfam: PF07238) was predicted by Amikam and Galperin by bioinformatics studies.54 This hypothesis was verified by Gomelsky and colleagues in the same year.55 The purified PilZ domain protein YcgR from E. coli showed a dissociation constant (Kd) of 0.84 ± 0.16 μM to c-di-GMP and the C terminus of BcsA from G. xylinus also binds to c-di-GMP.55 The PilZ domain is widely spread in many bacteria. Alg44 from P. aeruginosa,56 DgrA protein from C. crescentus,57 and PlzC and PlzD from V. cholerae58 are typical PilZ domain c-di-GMP receptors. The conserved sequence RxxxR20–30(D/N)x(S/A)xxG (x refers to any amino acid) in the PilZ domain, which is responsible for c-di-GMP binding, was disclosed by crystal structures and biochemical experiments.13,55,56
As stated earlier, DGC activity is allosterically inhibited when c-di-GMP binds to the I-site. c-di-GMP strongly inhibits PleD from C. crescentus with a Ki of 0.5 μM.59 A protein with degenerate GGDEF or EAL domains is another kind of c-di-GMP receptor. For example, the GGDEF–EAL domain FimX from P. aeruginosa has neither DGC nor PDE activity (PDB 3HV8).60 Its EAL domain binds to c-di-GMP and regulates twitching motility.60 Another example is the GGDEF domain of PopA from C. crescentus, which does not synthesize c-di-GMP but rather mediates movement toward the cell pole.61
Some c-di-GMP receptors are transcription factors that up-regulate or down-regulate target gene transcription upon c-di-GMP binding, such as the major flagella gene regulator in P. aeruginosa FleQ (PDB 4WXM).62 c-di-GMP binds to the Walker A motif of FleQ resulting in decreased flagella biosynthesis and increased EPS production.63 Furthermore, binding of c-di-GMP to VpsT, a transcription factor in V. cholerae, causes the dimerization of VpsT thereby enhancing its role in regulating biofilm formation.64
In 2008, Breaker and colleagues reported the first type of c-di-GMP riboswitch, Vc2 RNA.65 The riboswitch was observed to change conformation upon c-di-GMP binding. This facilitates the transcriptional regulation of downstream genes by the Vc2 riboswitch.65 The same group, in 2010, identified the c-di-GMP-II riboswitch which had picomolar affinity for c-di-GMP and was found to regulate self-splicing.66
Polymorphism of c-di-GMP
c-di-GMP has the tendency to readily form dimers, tetraplexes and higher aggregates in the presence of cations, not only in solution,67 but also when bound to proteins (see Fig. 8). Subtle changes in c-di-GMP can lead to a dramatic change in the propensity to form such higher aggregates.68 When developing c-di-GMP-based small molecule inhibitors against the various proteins and RNAs that bind to c-di-GMP, it is important to consider how any changes to the native c-di-GMP would affect the conformer and/or polyphorphism of the analog.68–70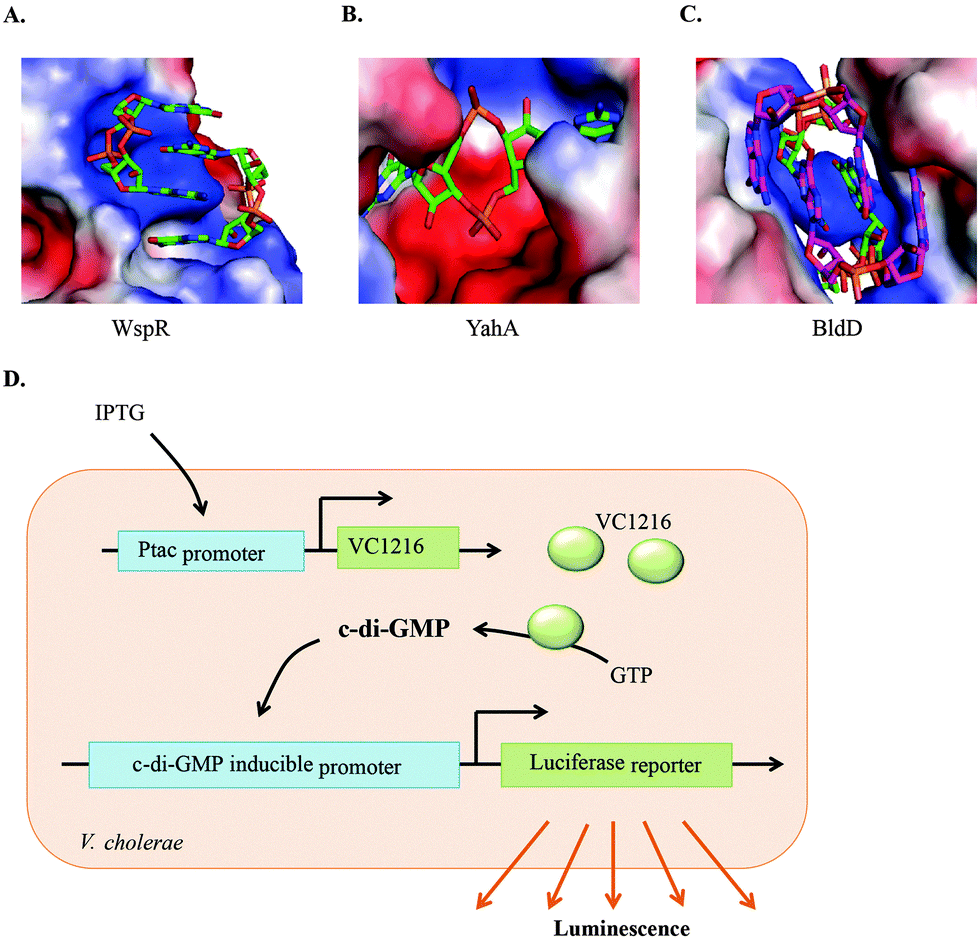 | ||
| Fig. 8 Polymorphism of c-di-GMP when bound to (A) the I-site of WspR, a DGC (PDB 3I5A), (B) YahA, PDE (PDB 4LJ3) and (C) BldD, a transcriptional factor (PDB 4OAZ). (D) Detection of c-di-GMP by V. cholerae luminescence.71 (A), (B), and (C) Reproduced with permission from ref. 72. Copyright © 2016 Future Science. | ||
Assays for the discovery of c-di-GMP inhibitors
Due to the importance of c-di-GMP signaling, there has been interest in developing small molecules to interrupt signaling. Towards this goal, several groups have developed assays that are amenable to high throughput screening inhibitor discovery. Following the discovery of c-di-GMP riboswitches that potentially bind to c-di-GMP, Sintim and colleagues reported a hybrid Spinach-Vc2 RNA aptamer, which couples the binding of c-di-GMP to the fluorescence of DFHBI,73 and this strategy has been utilized by others to develop riboswitch-based sensors that detect cyclic dinucleotides in complex environments, including inside bacterial cells.74–77 The riboswitch sensor, albeit being sensitive, is too expensive to use in high throughput screenings for inhibitor discovery. The Sintim group has demonstrated that intercalators that fluoresce or have fluorescence being quenched upon binding to c-di-GMP could be used as a simple means to screen for inhibitors of c-di-GMP processing enzymes. One example is that c-di-GMP can aggregate into supremolecular structures in the presence of thiazole orange (TO), leading to fluorescence turn-on.78 In another example, the same group demonstrated that c-di-GMP/proflavine association leads to fluorescence turn-off.79 A fluorescent c-di-NMP sensor that incorporates the aminopurine nucleobase and reports on PDE activity has also been reported.80 Such fluorescence assays could be used to screen for inhibitors of c-di-GMP synthesis or degradation. Others have used computational virtual screening81 and differential radial capillary action of ligand assay (DRaCALA)82 to identify c-di-GMP inhibitors. Perhaps a better way to discover c-di-GMP signaling inhibitors that are cell permeable is to use cell-based systems. Waters and colleagues have developed a V. cholerae bioluminescent reporter strain that can be used to screen for DGC inhibitors71 (see Fig. 8D). The reporter strain contains a plasmid harboring an IPTG-inducible DGC gene, VC1216. When treated with IPTG, VC1216 expression was induced with a corresponding increase in c-di-GMP production. The reporter strain also contains a second plasmid that encodes a luciferase operon under the control of a c-di-GMP inducible promoter. Any compound that inhibits VC1216 activity would lead to a change in bacterial luminescence. Such reporter strains or variations thereof are going to facilitate the discovery of cell-permeable c-di-GMP inhibitors.Inhibitors of c-di-GMP
The redundancy of GGDEF, EAL and HD-GYP domains in bacteria, which are typical domains of c-di-GMP DGCs and PDEs pose challenges for the development of small molecules to modulate c-di-GMP signaling in bacteria.83Table 1 shows the distribution of c-di-GMP DGCs and PDEs in select bacteria. c-di-GMP regulation by DGCs and PDEs is complicated at both the global level and the local level. Some DGCs and PDEs contribute to the total c-di-GMP concentration, but others only affect c-di-GMP concentration in a localized region.52 Römling and colleagues showed that in Salmonella, a mutation of the major DGC AdrA leads to a significant decrease in the global level of c-di-GMP.84 The expression of the biofilm regulator CsgD was not affected due to localized pools of c-di-GMP from two other DGCs STM2123 and STM3388.84 Some DGCs or PDEs regulate bacterial phenotypes without changing the overall c-di-GMP concentration. In Pseudomonas aeruginosa, mutating the nbdA gene which encodes the c-di-GMP PDE NbdA had no effect on intracellular c-di-GMP levels.85 However, the mutant strain was found to be resistant to biofilm dispersion by nitric oxide (NO) relative to the wildtype.85 Therefore a major challenge in inhibitor development is finding small molecules that can either inhibit all of these redundant enzymes or selectively inhibit one over the other.| Species | GGDEF | EAL | HD-GYP |
|---|---|---|---|
| Mesorhizobium loti | 32 | 18 | 1 |
| Pseudomonas aeruginosa | 33 | 21 | 3 |
| Escherichia coli | 19 | 17 | 0 |
| Bacillus subtilis | 4 | 3 | 0 |
| Bacillus halodurans | 4 | 2 | 2 |
| Mycobacterium tuberculosis | 1 | 2 | 0 |
| Vibrio cholerae | 41 | 22 | 9 |
| Caulobacter crescentus | 11 | 10 | 0 |
| Synechocystis sp. | 23 | 13 | 2 |
| Mycobacterium leprae | 3 | 2 | 0 |
| Xylella fastidiosa | 3 | 3 | 1 |
| Deinococcus radiodurans | 16 | 5 | 4 |
| Aquifex aeolicus | 11 | 8 | 1 |
So far, efforts to develop c-di-GMP signaling inhibitors have mainly been focused on finding inhibitors to interfere with c-di-GMP synthase DGC,82,86–93 PDE (EAL domain)94,95 and riboswitches.96,97 This is mostly because of the phenotypes that have been observed upon deletion of either DGC, PDE or their interacting partners. O'Toole and colleagues showed that deletion of the P. aeruginosa PDE, BifA, resulted in altered swarming mobility, EPS production and biofilm formation98 (Fig. 9). Also deletion of WspF, which caused the constitutive activation of the P. aeruginosa DGC WspR resulted in a strain with increased biofilm forming ability99 (Fig. 9). In a murine acute infection model, P. aeruginosa PA14 strains with mutation in the rocR gene were not virulent, evident by the survival of mice 5 days after infection100 (Fig. 9).
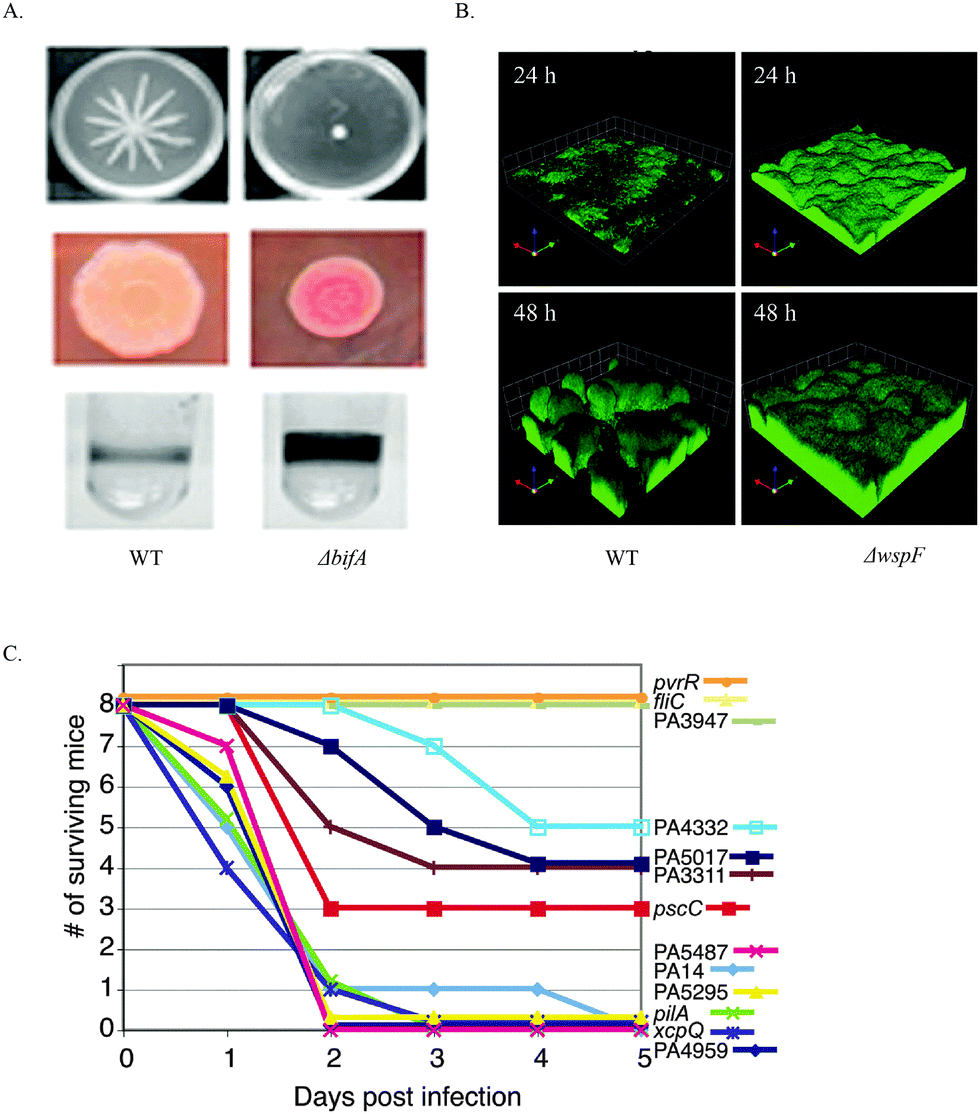 | ||
| Fig. 9 (A) The effects of c-di-GMP PDE BifA on swarming motility, EPS production and biofilm formation. BifA mutant attenuated swarming motility. Red, wrinkly colony on congo red plates indicated increased EPS production. BifA mutant strain biofilm formation was increased compared to WT strain. (B) WspF mutation resulted in the activation of WspR, a c-di-GMP DGC. ΔwspF strain produced a thicker biofilm than WT strain. (C) Survival of mice post infection with wildtype P. aeruginosa PA14 and strains with various mutations in the DGC, PDE and DGC–PDE genes. Strains with PA3947 gene (RocR) mutation are less virulent. (A) Reproduced with permission from ref. 98. Copyright © 2010 American Society for Microbiology. (B) Reproduced with permission from ref. 99. Copyright © 2005, The National Academy of Sciences of the USA. (C) Reproduced with permission from ref. 100. Copyright © 2006, The National Academy of Sciences of the USA. | ||
Table 2 and Fig. 10 depict the list of c-di-GMP inhibitors and their structures respectively. The first two examples of DGC inhibitors, glycosylated triterpenoid saponins (GTSs) and papulacandin B, were identified by Benziman and co-workers.93 However, such molecules probably lack the ability to cross bacterial membranes, making them not drug-like. Waters and co-workers reported several other small molecules, including N-(4-anilinophenyl)benzamide and LP-3145 as potential DGC inhibitors, to prevent biofilm formation in V. cholerae and P. aeruginosa respectively.87,88 Also, Landini and co-workers found that sulfathiazole and azathioprine inhibited biofilm formation by an E. coli strain harbouring P. aeruginosa DGC, WspR.89 The real mechanism is not known yet, however, sulfathiazole and azathioprine may affect the biosynthesis of c-di-GMP because of their antimetabolite activities, rather than directly inhibiting the activity of DGC.89,90 Of note, azathioprine is used as an anti-inflammatory drug in the treatment of Crohn's disease and rheumatoid arthritis.101 Therefore, its toxicity profile has already been established and could become a safe anti-biofilm drug for clinical applications. Recently, Lee and co-workers reported that ebselen inhibits WspR via covalent modification of a cysteine group near the active site.82 Ebselen is an alkylating agent as well as a planar aromatic molecule. It is also a promiscuous inhibitor and has been reported to inhibit many other biological targets102,103 and might not be an ideal drug candidate due to its potential toxicity.82 Others have also identified c-di-GMP signaling inhibitors via in silico screening. Rinaldo and co-workers identified catechol-containing sulfonohydrazide as an inhibitor of PleD with IC50 around 11 μM.81 From the foregoing, it is clear that the development of cell permeable inhibitors of c-di-GMP signaling is only at its infancy and many of the reported inhibitors do have various limitations that will prevent clinical development.
| Inhibitors | Functions | IC50 or Ki* (μM) | Ref. |
|---|---|---|---|
| * represents inhibitors for which Ki was determined | |||
| Nucleotide-based inhibitors | |||
| c-di-GMP | Allosterically inhibits c-di-GMP production | 48.9/15.42* | 92 |
| 2′-F-c-di-GMP | Inhibits DGC (WspR) | 11/12.6* | 92 |
| Inhibits PDE (RocR) | 0.7 | 92 | |
| Endo-S c-di-GMP | Inhibits PDE (RocR) | 0.14 | 95 |
| Inhibits PDE (RocR) | 0.02 | 92 | |
| 2′-H-c-di-GMP | Inhibits DGC (PleD) | 17.5 ± 1.1 | 108 |
| c-(RPRP)-di-Gps | Inhibits PDE (CC3396) | 0.48 | 94 |
| c-(RPSP)-di-Gps | Inhibits PDE (CC3396) | 0.82 | 94 |
| Neutral nucleotide-based inhibitors | |||
| DCI061 | Inhibits PDE (RocR) | 66.3 ± 1.3 | 108 |
| DCI058 | Inhibits DGC (PleD) | 25.5 ± 1.2 | 108 |
| Non-nucleotide-based inhibitors | |||
| Papulacandin B | Inhibits DGC (G. xylinum) | 70* | 93 |
| Glycosidic triterpenoid saponin (GTS) | Inhibits DGC (G. xylinun) | 5* | 93 |
| N-(4-Anilinophenyl)benzamide | Inhibits DGC (VC2370) | 1 | 87 |
| Inhibits DGC (WspR) | 17.8 | 87 | |
| LP-3134 | Inhibits DGC (WspR) and biofilm formation | 44.9 | 88 |
| LP-3145 | 70.9 | 88 | |
| LP-4010 | 102.4 | 88 | |
| LP-1062 | 73.1 | 88 | |
| Sulfathiazole | Inhibits c-di-GMP biosynthesis and biofilm formation | 5.8 | 90 |
| Azathioprine | Inhibits c-di-GMP biosynthesis and biofilm formation | 40 | 89 |
| Ebselen | Inhibits DGC (WspR) | 13.6 | 82 |
| Inhibits DGC (PleD) | 5 | 82 | |
| Amb2250085 | Inhibits DGC (PleD) | 11.05 | 81 |
| Amb379455 | Inhibits DGC (PleD) | 11.07 | 81 |
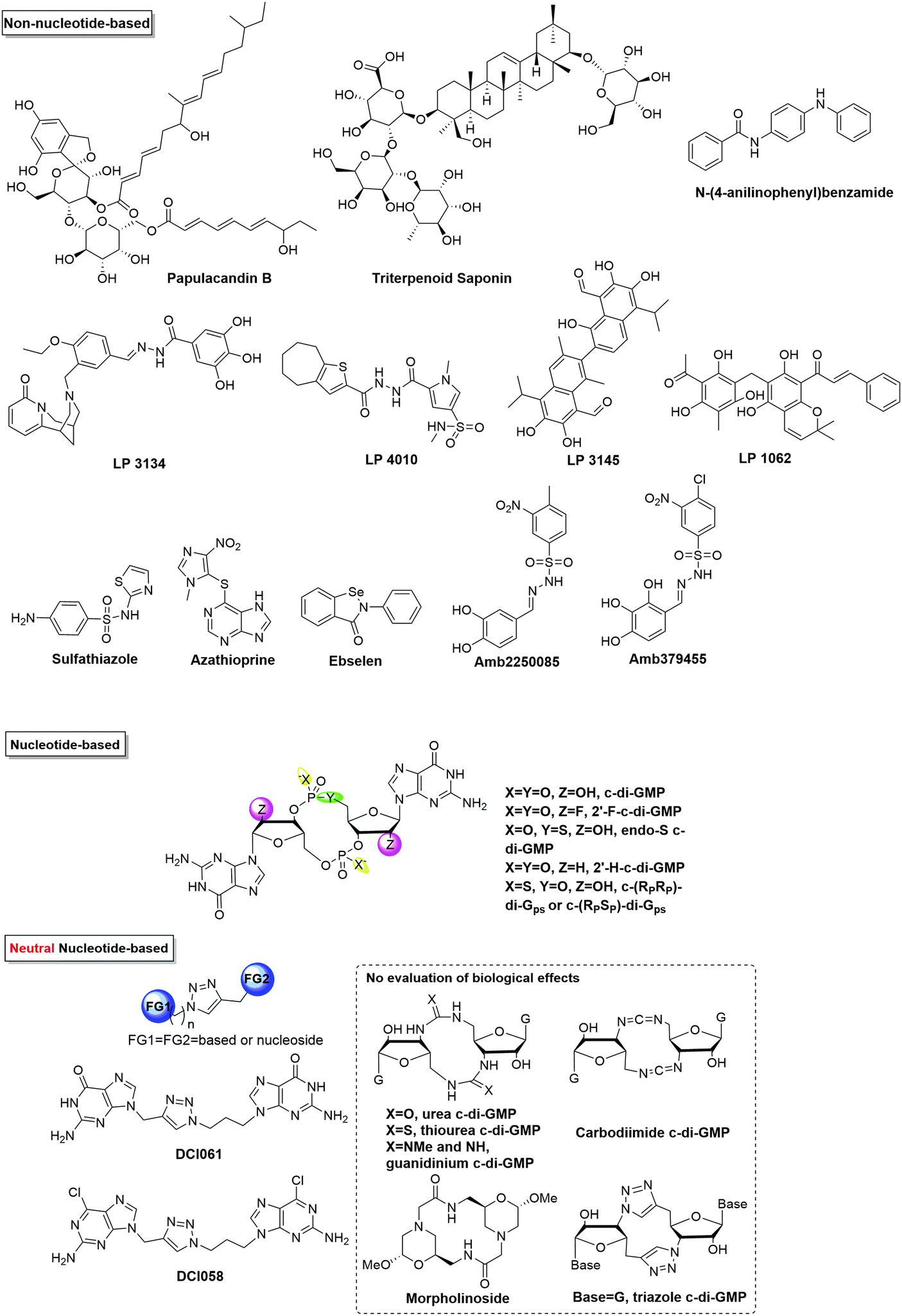 | ||
| Fig. 10 Structures of small molecule inhibitors of c-di-GMP. | ||
A number of nucleotide-based c-di-NMP analogs have been designed and synthesized in order to differentially inhibit different receptors, including backbone and sugar modifications of c-di-GMP53,68,92,94,104 and neutral analogs of c-di-GMP.105–108 Of note, endo-S c-di-GMP was shown to selectively inhibit c-di-GMP PDE but not DGC68 while 2′-F-c-di-GMP could inhibit DGCs better (4 times) than the native dinucleotide, c-di-GMP.92 Recently, Rinaldo and co-workers reported the first neutral c-di-GMP-like molecules for discriminating DGCs and PDEs, simply by replacing a phosphodiester bond with triazole.108 Potential limitations of c-di-GMP analogs as clinical candidates are solubility issues (guanine containing molecules tend to aggregate) and lack of cell permeation (most bisguanine-based analogs have too many hydrogen bond acceptor and donor units in the molecule and hence are not drug-like).
Recently, Sintim and co-workers have suggested an interesting strategy to inhibit c-di-GMP and c-di-AMP signaling via dinucleotide aggregation with small molecules (Fig. 11).72 Although only modest inhibitions of c-di-GMP/AMP degradation by PDEs were observed by this strategy, it is encouraging that one could bypass the enzyme redundancy issue that makes the global inhibition of c-di-NMP in bacteria challenging by targeting the dinucleotide itself.72
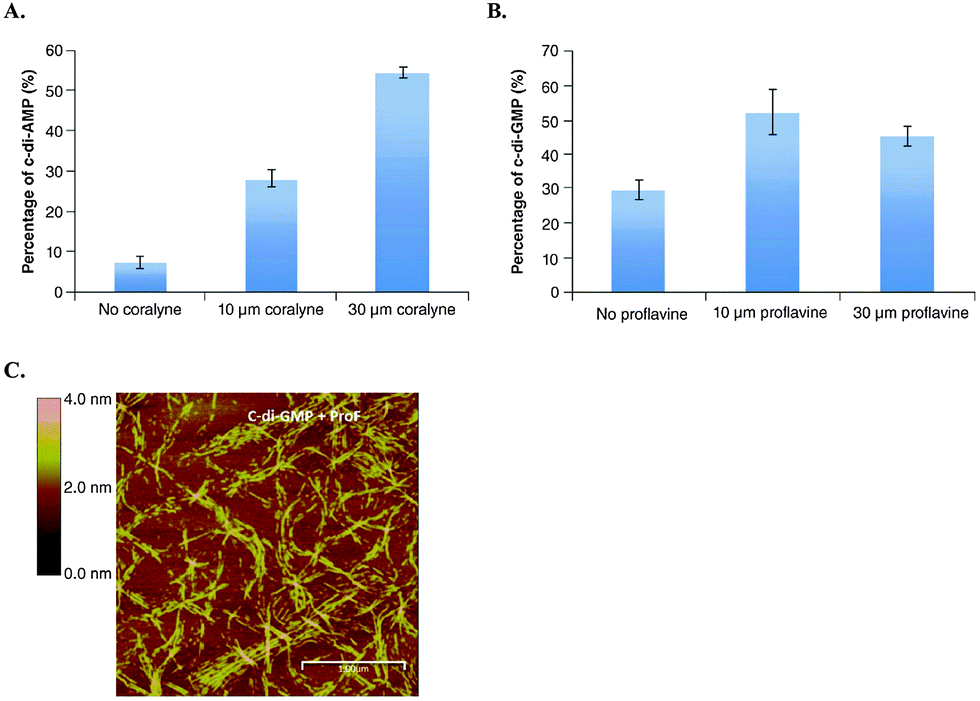 | ||
| Fig. 11 (A) YybT cleavage of 32P-c-di-AMP in the presence and absence of coralyne. (B) YybT cleavage of 32P-c-di-GMP in the presence and absence of proflavine. (C) Atomic force microscopy image of c-di-GMP proflavine aggregation. (A)–(C) Reproduced with permission from ref. 72 Copyright © 2016 Future Science. | ||
c-di-AMP
B. subtilis DNA integrity scanning protein A (DisA) is a protein involved in maintaining DNA integrity at the onset of B. subtilis sporulation.109,110 In the course of structural characterization of DisA from Thermatoga maritima, Hopfner and colleagues in 2008 discovered bis-(3′–5′) cyclic diadenylic acid (c-di-AMP) bound to the nucleotide binding site of DisA.109 The authors identified structural homologs of the nucleotide binding domain of DisA and hence predicted the existence and extensive role of c-di-AMP in archaea and eubacteria.109 Consistent with this, c-di-AMP signaling has been reported in several Gram-positive bacteria and mycobacteria including Staphylococcus aureus, L. monocytogenes, Mycobacterium tuberculosis, Streptococcus pyogenes and Streptococcus pneumoniae and in some Gram-negative bacteria such as Chlamydia trachomatis.111–115 Fluctuations in the intracellular concentration of c-di-AMP has been implicated in various physiological roles including fatty acid synthesis, potassium ion transport, cell wall homeostasis, host type I interferon response induction, and biofilm formation.112,116–119c-di-AMP metabolism enzymes
Very little is known about the environmental cues that elicit the production and degradation of c-di-AMP. Also the direct relationship between cellular c-di-AMP levels and the phenotypes observed remains to be elucidated. For example, low c-di-AMP levels sensitize B. subtilis and L. monocytogenes to cell wall targeting antibiotics.118,122 In S. aureus, high cellular c-di-AMP levels result in increased peptidoglycan crosslinking and resistance to cell wall targeting antibiotics.111 High c-di-AMP levels have also been shown to trigger stringent response resulting in an increase in the levels of the alarmone (p)ppGpp in S. aureus.18 Exogenously added c-di-AMP enhanced B. subtilis sporulation120 while overexpression of MsDisA in M. smegmatis (implying increased c-di-AMP levels) resulted in minute colonies121 (Fig. 12). Consequently, the cellular concentration of c-di-AMP is tightly regulated.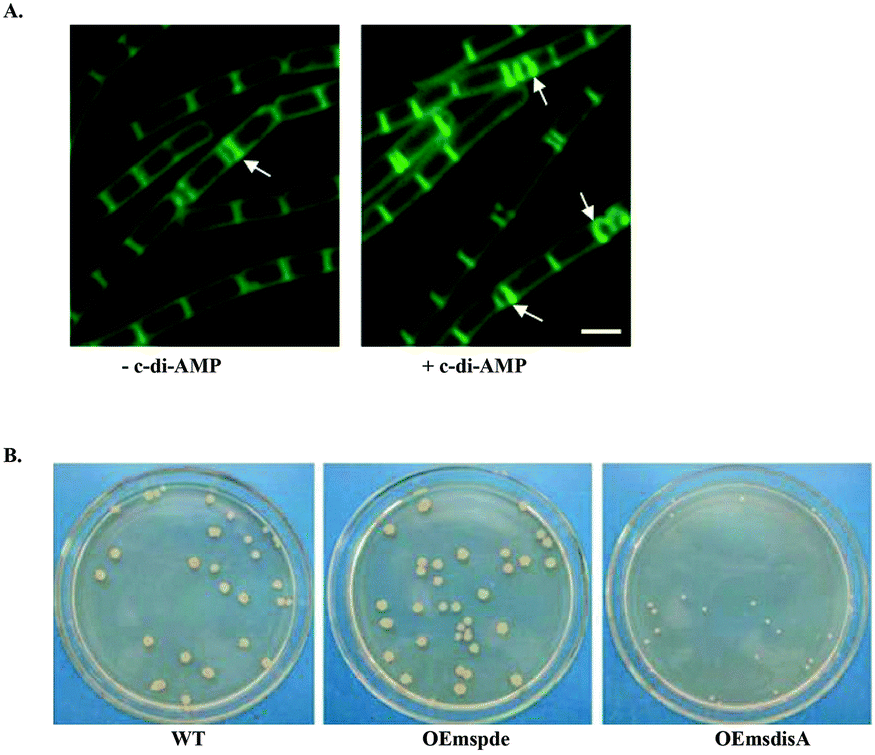 | ||
| Fig. 12 (A) Addition of exogenous c-di-AMP (+ c-di-AMP) to B. subtilis cells showed an increase in sporulation characterized by the higher number of polar septa (white arrows) observed. (B) Overexpression of the DAC, DisA in M. smegmatis results in minute colonies compared to WT and PDE overexpression strains. (A) Reproduced with permission from ref. 120 © (2011) John Wiley & Sons. (B) Reproduced with permission from ref. 121. | ||
c-di-AMP is synthesized at the dimer interface of DisA_N (Pfam PF02457) domain-containing enzymes known as diadenylate cyclases (DAC)26 (Fig. 13 and 15). The active pocket contains two metal ions, which coordinate the phosphate moieties of two ATP molecules. A condensation of the two ATP molecules by active site residues forms c-di-AMP (Fig. 13).109,123 The DAC domain is conserved among the bacteria that utilize c-di-AMP signaling and it is characterized by a catalytic site with the conserved RHR (Arg-His-Arg) and DGA (Asp-Gly-Ala) motifs.109,123 Such enzymes have been reported in numerous bacteria including B. subtilis (DisA,109 CdaA and CdaS124), S. pyogenes (CdaA),125S. pneumoniae (CdaA),126M. tuberculosis (MtDisA),113C. trachomatis (CdaA)115 and L. monocytogenes (CdaA; PDB 4RV7).127 DAC enzymes increase the local and/or global pools of c-di-AMP in bacteria. Attempts to delete the DAC gene from bacteria such as the human pathogen L. monocytogenes have been unsuccessful;112 pointing to the essentiality of c-di-AMP signaling in bacterial growth and physiology. Consequently, DAC has been viewed as a potential target for antibiotic development.
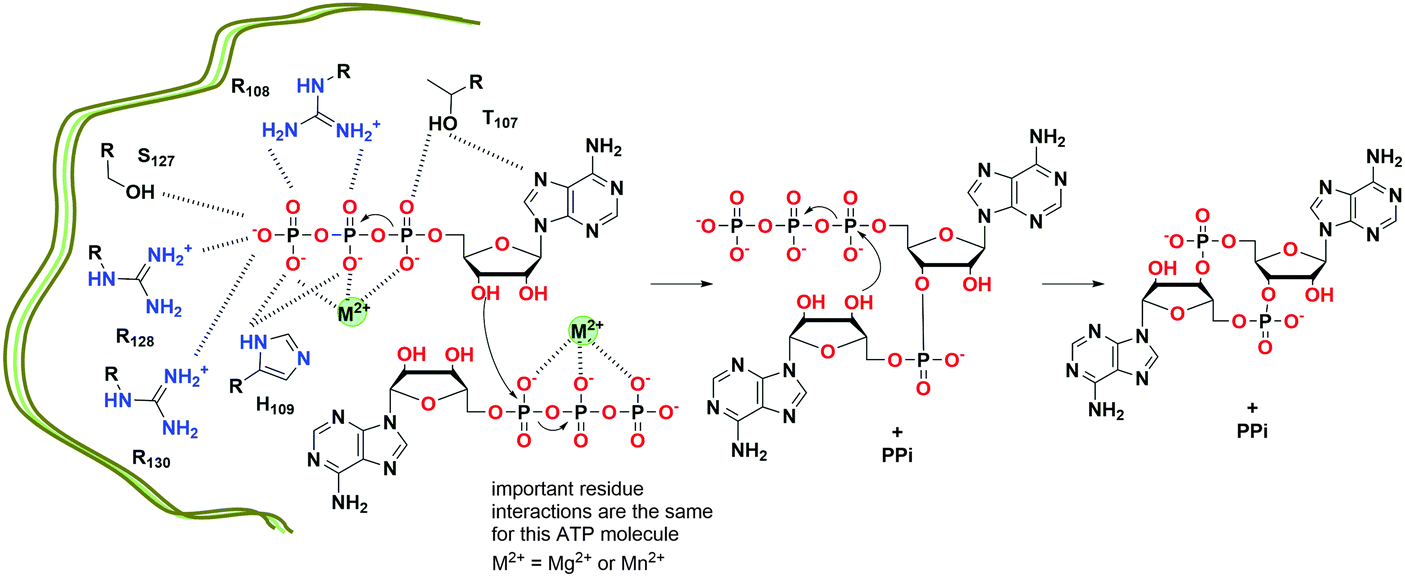 | ||
| Fig. 13 Proposed mechanism of c-di-AMP formation by T. maritima DisA.128 (based on PDB 4YVZ and 4YXJ). | ||
After effecting a signaling process or at higher than normal cellular concentrations, c-di-AMP is degraded by phosphodiesterase (PDE) to the linear pApA,17 a process which requires two metal ions129 (Fig. 14). Three families of phosphodiesterases have been identified to degrade c-di-AMP. Liang and co-workers identified B. subtilis GdpP (GGDEF domain protein-containing phosphodiesterase), belonging to the DHH-DHHA1 family of PDEs, as the first c-di-AMP PDE.129 The domain architecture of GdpP and its homologs are characterized by two transmembrane helical domains, the PAS (Per-Arnt-Sim) domain, a degenerate GGDEF domain and the DHH-DHHA1 catalytic domains. This family of PDEs has been shown in other bacteria such as S. aureus (GdpP)111 and L. monocytogenes (PdeA).118 A related family of PDEs is those that contain only the catalytic DHH-DHHA1 domain as found in B. burgdorferi (DhhP),130S. pneumoniae (Pde1 and Pde2)126 and M. tuberculosis (CnpB).131,132 A third family of PDEs which belongs to the 7TMR-HD family was identified by Woodward and coworkers.133 The PDE, PgpH which was identified in L. monocytogenes, possesses a catalytic histidine-aspartate (HD) domain.133 For a comprehensive overview of c-di-AMP PDEs, the reader is referred to a recent review by Huynh and Woodward.17 Another approach to decreasing intracellular c-di-AMP concentration could be seen in intracellular bacteria such as L. monocytogenes, C. trachomatis and M. tuberculosis.112,115,132 During infection, these pathogens secrete c-di-AMP into the host cytosol which induces type I IFN response (Fig. 15). Overall, DACs, PDEs and export proteins work together to maintain c-di-AMP homeostasis (Fig. 15).
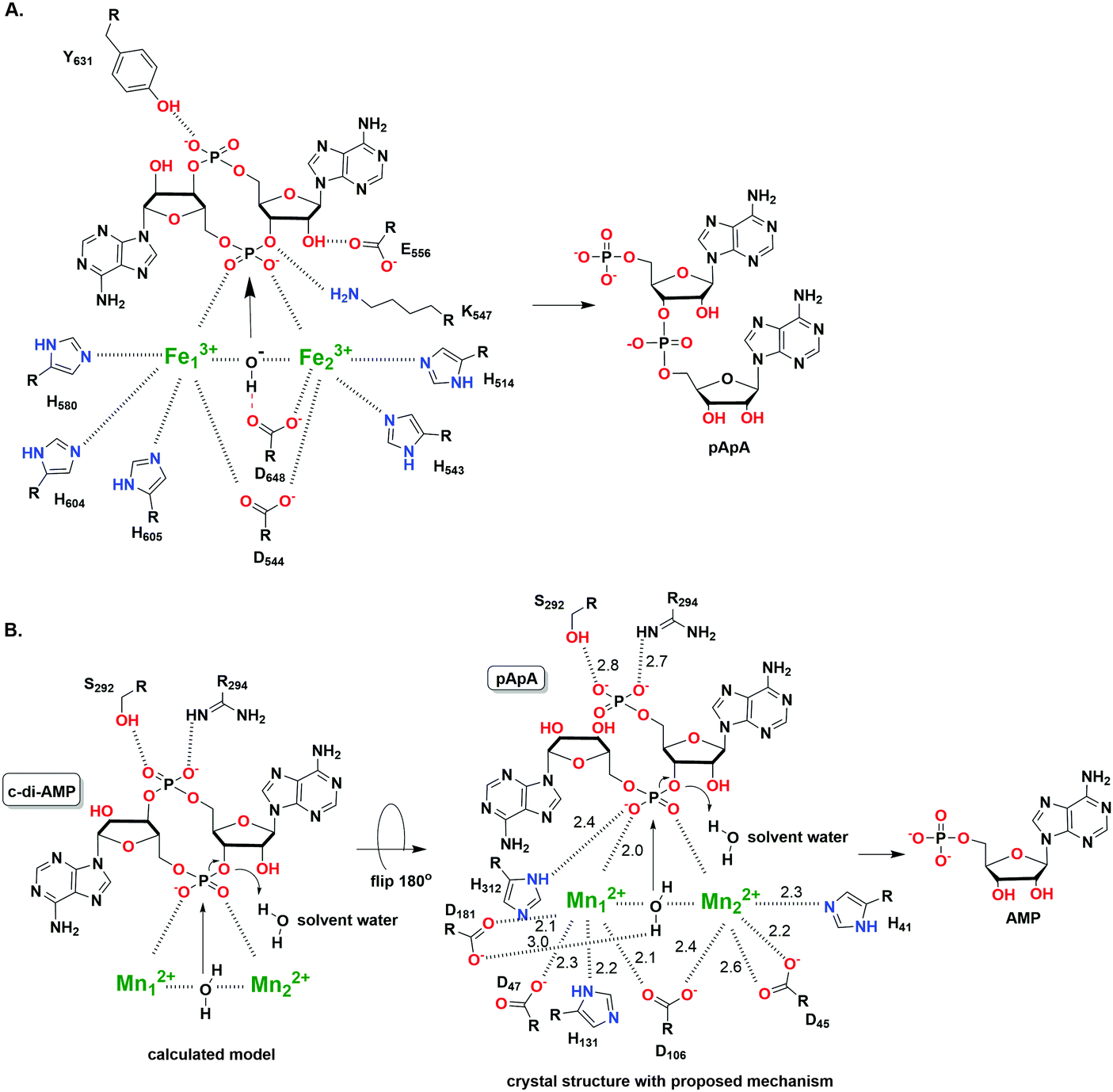 | ||
| Fig. 14 (A) The hydrolysis mechanism of c-di-AMP by the HD domain phosphodiesterase from Listeria monocytogenes, PgpH.133 (PDB 4S1B). (B) The proposed hydrolysis mechanism of pApA by DHH-DHHA1 domain phosphodiesterase from Mycobacterium tuberculosis (Rv2837c).134 (PDB 5CEU). (A) Adapted with permission from ref. 133 Copyright © 2015, National Academy of Sciences, USA (B) Adapted with permission from ref. 134. Copyright © 2016, The American Society for Biochemistry and Molecular Biology. | ||
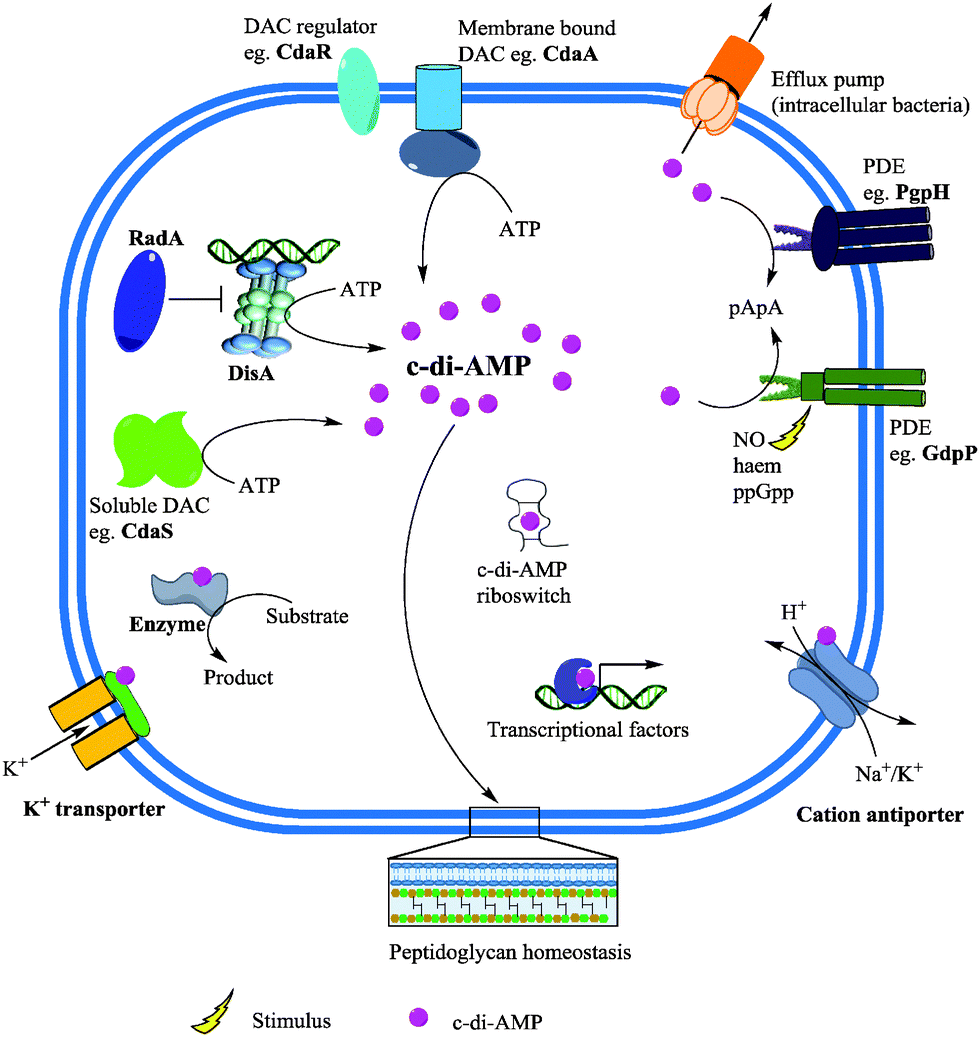 | ||
| Fig. 15 c-di-AMP metabolism and effector functions. Membrane bound and/or soluble DACs synthesize c-di-AMP from ATP. c-di-AMP binds to receptor/target proteins and RNA to modulate physiological functions. Phosphodiesterases (PDE) degrade c-di-AMP into the linear pApA or AMP and the second messenger could also be secreted by the efflux pumps of some intracellular bacteria. Adapted from ref. 135 Copyright © 2015 Zheng, Ma, Wang, Xie, Ali and He. | ||
c-di-AMP receptors
Binding of second messengers is a crucial step in signal transduction. After the discovery of c-di-AMP, a significant amount of effort was directed towards the identification of macromolecules that respond to fluctuations in cellular c-di-AMP concentration by binding to the second messenger. Several c-di-AMP binding partners have been identified encompassing proteins and RNA riboswitches (Fig. 15). Zhang et al. discovered the first c-di-AMP binding protein DarR, a TetR family transcription factor in M. smegmatis.116 The authors noted that the DNA binding activity of DarR was enhanced after binding c-di-AMP which results in the repression of expression of various genes, including genes associated with metabolism and transportation of fatty acids.116 The c-di-AMP binding proteins KtrA in S. aureus136 and CabP in S. pneumoniae117 enable bacterial survival under low-potassium conditions and elevated c-di-AMP levels have been shown to inhibit potassium uptake. KdpD and CpaA which are S. aureus proteins predicted to be involved in potassium homeostasis and cation–proton antiporting respectively, have c-di-AMP binding sites.136 Also, the PII-like signal transduction proteins B. subtilis DarA137 and S. aureus PstA138 have been demonstrated to possess c-di-AMP binding affinity. L. monocytogenes pyruvate carboxylase (LmPC), which is essential for the growth of the human pathogen, was also shown to bind c-di-AMP. Binding of c-di-AMP was observed to inhibit the activity of LmPC.139 The ydaO riboswitch class found in several bacteria has also been shown to have strong affinity for c-di-AMP140 compared to ATP which was earlier shown to be a ligand for the riboswitch.141Inhibitors of c-di-AMP
c-di-AMP is undoubtedly an important second messenger in Gram-positive bacteria and the essentiality of c-di-AMP synthases has spurred the development of DAC inhibitors. c-di-AMP detection assays could facilitate inhibitor discovery. Bai and colleagues developed an ELISA-based assay for the detection and quantification of c-di-AMP using the c-di-AMP binding protein, CabP from S. pneumoniae.142 The assay was used to identify some c-di-AMP binding proteins.142 The Sintim group has also developed a surprisingly simple assay that uses commercially available coralyne to detect c-di-AMP and the group has used this assay to identify various inhibitors of DisA (Fig. 16).143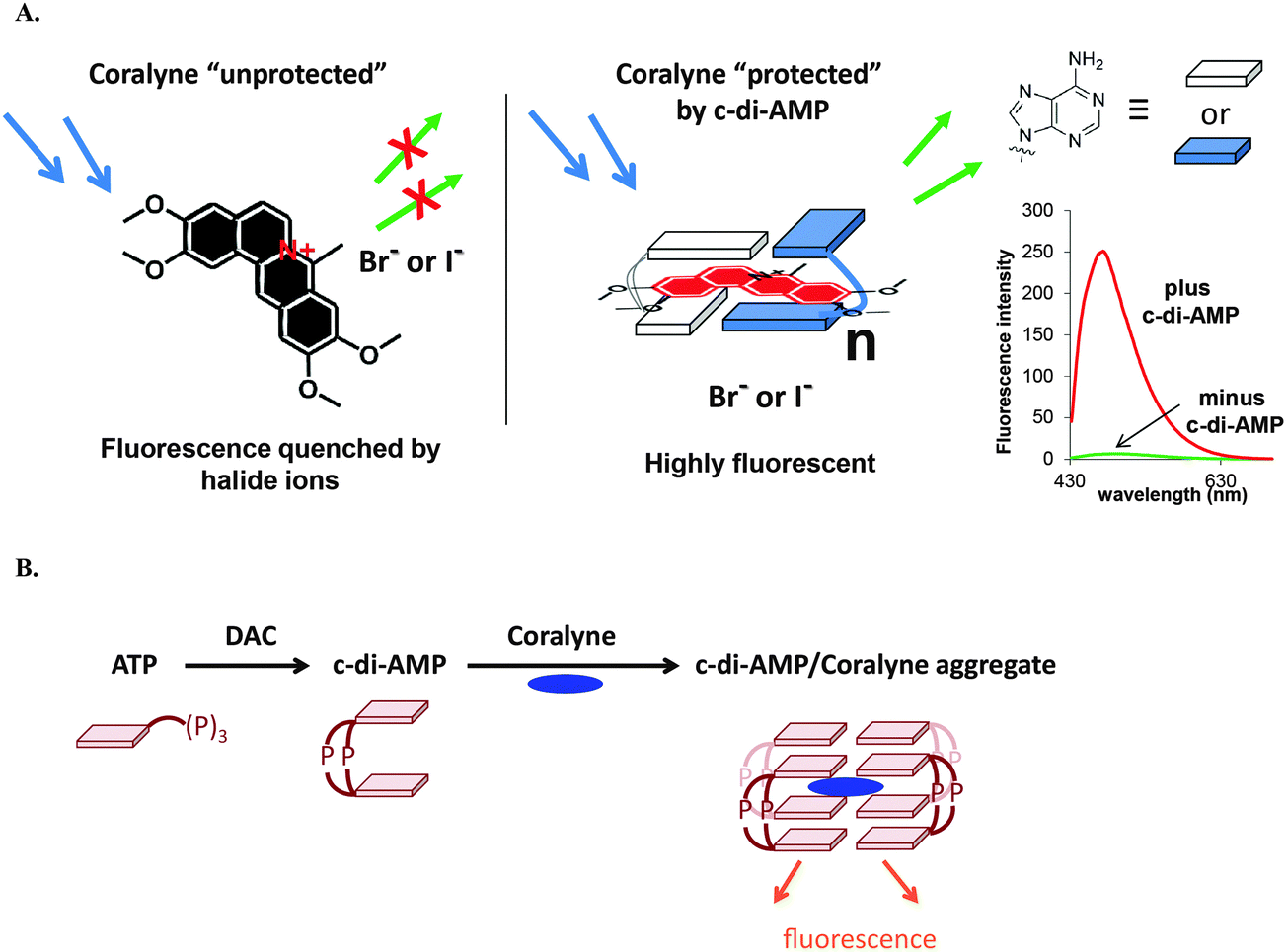 | ||
| Fig. 16 (A) The principle of the coralyne fluorescence assay for c-di-AMP detection. (B) Schematic illustration of a DAC reaction showing the synthesis of c-di-AMP and the fluorescent c-di-AMP/coralyne complex. (A) Reproduced with permission from ref. 143 Copyright © 2014, American Chemical Society. (B) Reproduced with permission from ref. 144 Copyright © 2014, The Royal Society of Chemistry. | ||
Cyclic di-AMP as stated earlier regulates cell wall homeostasis. With a majority of antibiotics targeting the bacterial cell wall, it is possible that inhibitors of c-di-AMP signaling could potentiate the activities of traditional cell wall-targeting antibiotics. However, there is a paucity of inhibitors against c-di-AMP metabolism enzymes129,144 (Fig. 17 and Table 3). Sintim and co-workers established a high-throughput fluorescence assay for c-di-AMP detection using the coralyne fluorophore.143 After screening several compound libraries, three inhibitors of B. subtilis DisA were identified.144–146 Bromophenol-TH was the first documented DAC inhibitor. It was shown to specifically inhibit DisA with an IC50 of 56 μM.117 Suramin, an antiparasitic drug, and the tea polyphenol theaflavin digallate were also identified as potent DisA inhibitors with IC50 of 1.1 μM and 3.4 μM145,146 respectively. Witte and co-workers also reported that 3′-deoxyATP inhibited Thermotoga maritima DisA with a IC50 of 3.8 μM.123 Liang and co-workers have shown that the stringent stress alarmone ppGpp could competitively inhibit B. subtilis GdpP (formerly YybT), with a Ki of 35.9 ± 7.2 μM.129 Additionally, ppGpp also inhibits GdpP in S. aureus with a Ki of 129.7 ± 42.8 μM18 and PgpH in L. monocytogenes with a IC50 of 200–400 μM.133 Like c-di-GMP, the structure–activity relationship of c-di-AMP was also thoroughly studied by Strobel et al. and some of those analogs have been identified to be resistant to GdpP cleavage.147
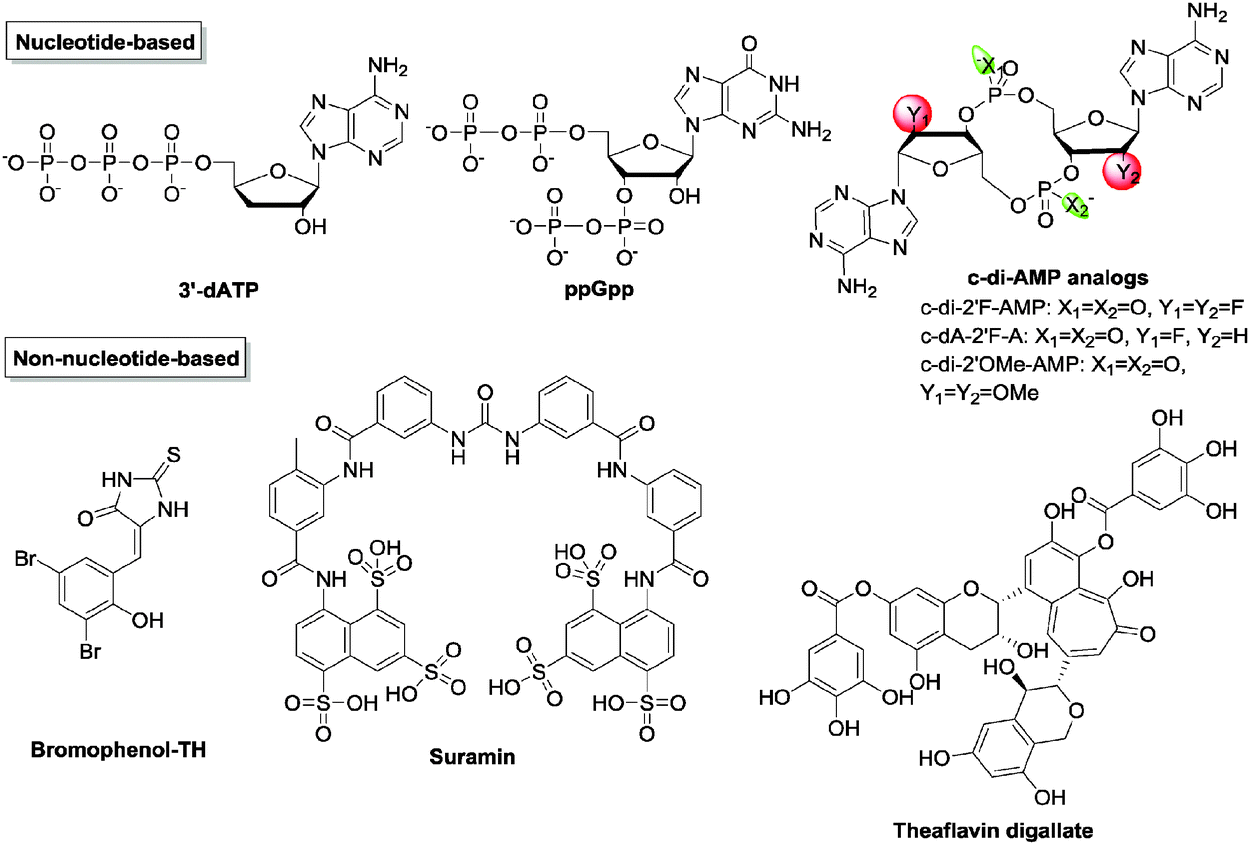 | ||
| Fig. 17 Structures of representative c-di-AMP inhibitors. | ||
| Inhibitors | Functions | IC50 (μM) | Ref. |
|---|---|---|---|
| Nucleotide-based inhibitors | |||
| 3′-dATP | Inhibits DAC (DisA) | 3 | 123 |
| ppGpp | Inhibits PDE (YybT) | 35.9 ± 7.2 | 129 |
| Inhibits PDE (GdpP) | 129.7 ± 42.8 | 18 | |
| Inhibits PDE (PgpH) | 200–400 | 133 | |
| c-di-2′F-AMP | Inhibits PDE (GdpP) | 26 ± 3.6 | 147 |
| c-dA-2′F-A | Inhibits PDE (GdpP) | 9.4 ± 0.8 | 147 |
| c-di-2′OMe-AMP | Inhibits PDE (GdpP) | 15 ± 2.5 | 147 |
| Non-nucleotide-based inhibitors | |||
| Bromophenol-TH | Inhibits DAC (DisA) | 56 | 148 |
| Suramin | Inhibits DAC (DisA) | 1.1 | 145 |
| Theaflavin digallate | Inhibits DAC (DisA) | 3.4 | 146 |
3′3′-cGAMP
In 2012, Mekalanos and colleagues explored the contribution of the Vibrio seventh pandemic island-1 (VSP-1) to pathogenesis and they discovered a new type of cyclic dinucleotide with hybrid bases, cyclic AMP–GMP (3′3′-cGAMP), in V. cholerae.22 This new bacterial second messenger is synthesized by a novel class of dinucleotide cyclase DncV,22 which contains a conserved G[G/S]X9–13DX[D/E] motif. DncV represses V. cholerae chemotaxis, a requirement for intestinal colonization.22 Subsequently, Jiang and colleagues reported three cGAMP-specific PDEs in V. cholerae with HD-GYP domains, named V-cGAP1/2/3.149 All of three PDEs were able to degrade 3′3′-cGAMP to the linear 5′-pApG but the 5′-nucleotidase activity of V-cGAP1 afforded it the capacity to cleave 5′-pApG to 5′-ApG in a second step.149 The protein receptors for 3′3′-cGAMP are still unknown. The first 3′3′-cGAMP riboswitch was developed based on the class I c-di-GMP riboswitch. In 2011, Strobel and colleagues found that a single C92U mutation in the ligand binding pocket enables the c-di-GMP class I riboswitch to bind to 3′3′-cGAMP.150 In 2015, Hammond showed that Gram-negative bacterium Geobacter sulfurreducens produced 3′3′-cGAMP and that the GEMM-I (Genes for the Environment, Membranes, and Motility) class riboswitch was a 3′3′-cGAMP receptor that regulates electrophysiology genes.151 At the same time, Breaker and colleagues also reported the 3′3′-cGAMP riboswitch in Deltaproteobacteria that controls exoelectrogenesis.1522′3′-cGAMP
In 2013, a cyclic dinucleotide second messenger, 2′3′-cGAMP present mammals was uncovered by Ablasser, Hornung and colleagues.23 One of the two phosphodiester bonds has a special linkage between 2′-OH of GMP and 5′-phosphate of AMP, distinguishing the mammalian 2′3′-cGAMP from other bacterial cyclic dinucleotides.153 2′3′-cGAMP is synthesized from GTP and ATP by a cytoplasmic nucleotidyl transferase known as cyclic GMP–AMP synthase (cGAS).23 The 2′3′-cGAMP synthesis was postulated to occur in two steps. In a first step, GTP and ATP form the linear intermediate pppGp(2′–5′)A which is cyclized to 2′3′-cGAMP in the second step.154 The enzymatic activity of cGAS is stimulated by double stranded DNA in the cytoplasm. When cGAS binds to cytoplasmic DNA, it synthesizes the signaling molecule 2′3′-cGAMP which induces immune response.155,156 In 2014, Mitchison and colleagues identified a glycoprotein on the plasma membrane and endoplasmic reticulum, ENPP1 that degraded 2′3′-cGAMP into AMP and GMP (kcat = 12 s−1, Km = 20 μM).157 ENPP1 was originally reported to be an ATP hydrolase (kcat = 16 s−1, Km = 46 μM). The crystal structure of ENPP1 (PDB 4HTW) revealed a Ca2+-binding domain and a site for two Zn2+ ions in its active pocket.158Type I interferon response
One of the efficient methods for the host innate immune system to detect intracellular pathogens is to sense cytoplasmic DNA. As described previously, upon binding to cytoplasmic double stranded DNA, cGAS synthesizes the signaling molecule 2′3′-cGAMP.159 2′3′-cGAMP binds to and activates STING (stimulator of interferon genes, also known as MITA, MPYS and ERIS), which is a 5 transmembrane domain protein that predominantly resides in the endoplasmic reticulum.23 Activated STING triggers the phosphorylation of transcriptional factor IFN regulatory factor (IRF3) by the kinase TANK-Binding Kinase 1 (TBK1).160,161 Phosphorylated IRF3 then translocates into the nucleus to induce the transcription of type I interferon genes (Fig. 18). The affinity of 2′3′-cGAMP for human STING has been shown to be very high, with a dissociation constant of 4.59 nM.153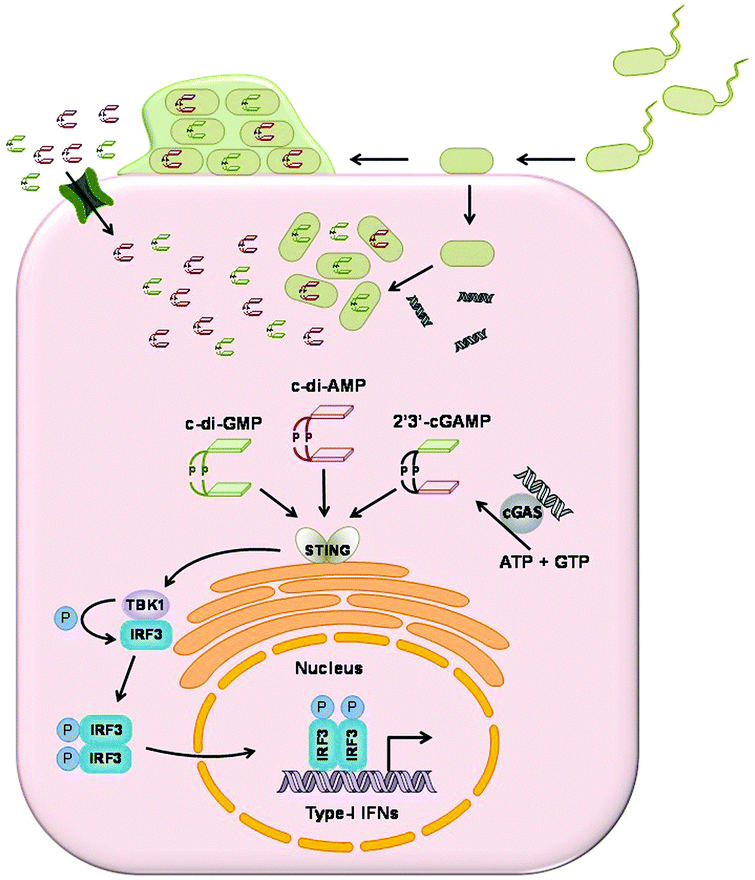 | ||
| Fig. 18 Cyclic dinucleotides induce type I interferon response. cGAS detects cytoplasmic DNA and synthesizes 2′3′-cGAMP. 2′3′-cGAMP and c-di-AMP and c-di-GMP from bacteria bind to STING. Activated STING mediates the phosphorylation of IRF3 by TBK1. Phosphorylated IRF3 induces the transcription of type I interferon genes in the nucleus. | ||
STING is able to sense cytoplasmic DNA directly,162 as well as the bacterial cyclic dinucleotides.163 Vance and colleagues showed the direct binding of STING to c-di-GMP and they obtained a Kd of 5 μM by equilibrium dialysis.163 c-di-AMP is capable of inducing type I interferon response in a STING-dependent manner but with a lower binding affinity.115 Chen and colleagues measured the binding affinity of STING to 3′3-cGAMP and synthetic analogs 3′2′-cGAMP and 2′2′-cGAMP by ITC. Kd was measured as 1.04 μM, 1.61 μM and 287 nM. Their binding affinities are much lower than the host derived 2′3′-cGAMP.153
Small molecules that inhibit bacterial or human cyclic dinucleotide PDEs to increase cytosolic c-di-NMP in immune cells or activate cGAS to produce 2′3′-cGAMP or bind directly to STING to activate immune response could have anti-virulence properties. These molecules could however lead to unwanted prolonged inflammation and its associated unwanted effects such as pain, cancer and so on. On the other hand, it is also possible to develop c-di-NMP analogs that would bind to STING but prevent STING-related interferon response. Such molecules could have anti-inflammatory properties.
Cyclic dinucleotides as anticancer agents or cancer immune adjuvants
c-di-GMP has been shown to possess broad activities, such as inhibiting bacterial infection,164 stimulating host immune response,165 acting as an adjuvant for vaccination165 and inhibiting cancer cell proliferation.166 The potential of c-di-GMP as an agent against metastatic breast cancer, using a mice tumor model, has recently been demonstrated by Gravekamp.167 In this study, Gravekamp and co-workers firstly infected mice with metastatic breast cancer and then immunized them with an attenuated L. monocytogenes (LM)-based vaccine. After 19 days of treatment with low doses of c-di-GMP, the mice were able to survive with almost no metastases observed as well as improved T-cell responses. When treating mice infected with metastatic breast cancer by using one high dose and subsequent low doses of c-di-GMP, without the LM-based vaccine, they could achieve equal effectiveness against metastases.167 The real mechanism has not been fully elucidated. One possibility is that the LM-based vaccine infects myeloid-derived suppressor cells (MDSC) and c-di-GMP activates T cells, both toward reducing immune suppression and improving therapy. They also proposed that in the absence of the LM-based vaccine, high-dose of c-di-GMP would induce tumor cell death, while low-dose of c-di-GMP would induce activation of T-cells.167Karaolis et al., in 2007, proposed that cyclic dinucleotides like c-di-GMP could be used as immunotherapeutic candidates in both humans and animals.165 However, the two phosphate groups on c-di-GMP inevitably prevents it from being developed as a drug candidate since it can barely cross cell membranes and may also be easily hydrolyzed.168 To address such issues, several groups have shown how to generate c-di-GMP inside host cells or transport c-di-GMP into mammalian cells for immunotherapy.168–172 Waters and co-workers used the adenovirus serotype 5 (Ad5) vector to transduce dgc into mammalian cells, and thus increase intracellular c-di-GMP.169 In a more recent study, they combined VCA0848 (more potent dgc) with nonreplicating adenovirus serotype 5 (AdVCA0848) for an improved stimulation of innate immunity in mice.170 On the other hand, the Gursel group used the arginine peptide (positively charged) to complex with c-di-GMP (negatively charged) for delivery into mouse cells with enhanced immunostimulation of c-di-GMP being observed.168 In addition, Hayakawa loaded c-di-GMP on liposomes (YSK05 lipid), facilitating the delivery of c-di-GMP directly into the cytosol. As a result, c-di-GMP enhanced anticancer activity against malignant melanomas.171,172
Recently and unsurprisingly 2′3′-cGAMP has been identified as an immune adjuvant in mouse studies.173 Replacement of phosphodiester linkage by the phosphothioate bond resulted in a 2′3′-cGAMP analog resistant to hydrolysis while capable of activating human STING.157 Though such a 2′3′-cGAMP analog still possesses negative charges, it opens up the field to develop hSTING agonists as cancer immunotherapeutic drugs.
Conclusions
The importance of the physiological roles played by cyclic dinucleotides is well established. As a result significant effort has been directed towards identifying receptor/target proteins and RNA. The use of chemical probes represents one obvious approach to studying the molecular mechanisms underlying the phenotypes observed as a result of cyclic dinucleotide signaling. However, thus far only a handful of inhibitors have been uncovered for c-di-GMP and c-di-AMP metabolism. Furthermore, there are no inhibitors documented for the hybrid cyclic dinucleotide cGAMP (2′3′ and 3′3′) signaling. We believe that c-di-NMP signaling is on par with cNMP signaling or protein kinase signaling. Considering that inhibitors of these two signalings have had a tremendous impact on modern medicine, it is safe to predict that inhibitors of c-di-NMP signaling would also positively impact clinical practice. We hope that this review provides a rallying cry to other medicinal chemists to join the cyclic dinucleotide revolution and develop inhibitors against the various signaling pathways mediated by these fascinating molecules.Acknowledgements
We thank the NSF (CHE grants 0746446 and 1307218) for funding our cyclic dinucleotide projects.Notes and references
- D. Kalia, G. Merey, S. Nakayama, Y. Zheng, J. Zhou, Y. Luo, M. Guo, B. T. Roembke and H. O. Sintim, Chem. Soc. Rev., 2013, 42, 305–341 RSC.
- M. Gomelsky, Mol. Microbiol., 2011, 79, 562–565 CrossRef CAS PubMed.
- K. A. Lucas, G. M. Pitari, S. Kazerounian, I. Ruiz-Stewart, J. Park, S. Schulz, K. P. Chepenik and S. A. Waldman, Pharmacol. Rev., 2000, 52, 375–414 CAS.
- J. N. Marden, Q. Dong, S. Roychowdhury, J. E. Berleman and C. E. Bauer, Mol. Microbiol., 2011, 79, 600–615 CrossRef CAS PubMed.
- C. Pesavento and R. Hengge, Curr. Opin. Microbiol., 2009, 12, 170–176 CrossRef CAS PubMed.
- V. Hauryliuk, G. C. Atkinson, K. S. Murakami, T. Tenson and K. Gerdes, Nat. Rev. Microbiol., 2015, 13, 298–309 CrossRef CAS PubMed.
- I. M. Ernst, R. Fliegert and A. H. Guse, Front. Immunol., 2013, 4, 259 CAS.
- X. Gu, Z. Yang, L. Zhang, S. Kunerth, R. Fliegert, K. Weber and A. H. Guse, J. Med. Chem., 2004, 47, 5674–5682 CrossRef CAS PubMed.
- P. C. Lee, B. R. Bochner and B. N. Ames, J. Biol. Chem., 1983, 258, 6827–6834 CAS.
- A. Varshavsky, Cell, 1983, 34, 711–712 CrossRef CAS PubMed.
- L. L. Kisselev, J. Justesen, A. D. Wolfson and L. Y. Frolova, FEBS Lett., 1998, 427, 157–163 CrossRef CAS PubMed.
- R. Hengge, A. Gründling, U. Jenal, R. Ryan and F. Yildiz, J. Bacteriol., 2016, 198, 15–26 CrossRef CAS PubMed.
- U. Römling, M. Y. Galperin and M. Gomelsky, Microbiol. Mol. Biol. Rev., 2013, 77, 1–52 CrossRef PubMed.
- T. H. Pham, Z. X. Liang, E. Marcellin and M. S. Turner, Curr. Genet., 2016, 1–8 Search PubMed.
- Z. X. Liang, Nat. Prod. Rep., 2015, 32, 663–683 RSC.
- D. G. Ha and G. A. O'Toole, Microbiol. Spectrum, 2015, 3, MB-0003-2014 Search PubMed.
- T. N. Huynh and J. J. Woodward, Curr. Opin. Microbiol., 2016, 30, 22–29 CrossRef CAS PubMed.
- R. M. Corrigan, L. Bowman, A. R. Willis, V. Kaever and A. Gründling, J. Biol. Chem., 2015, 290, 5826–5839 CrossRef CAS PubMed.
- D. Gao, T. Li, X. D. Li, X. Chen, Q. Z. Li, M. Wight-Carter and Z. J. Chen, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E5699–E5705 CrossRef CAS PubMed.
- P. Ross, H. Weinhouse, Y. Aloni, D. Michaeli, P. Weinbergerohana, R. Mayer, S. Braun, E. Devroom, G. A. Vandermarel, J. H. Vanboom and M. Benziman, Nature, 1987, 325, 279–281 CrossRef CAS PubMed.
- U. Römling, Sci. Signaling, 2008, 1, pe39 CrossRef PubMed.
- B. W. Davies, R. W. Bogard, T. S. Young and J. J. Mekalanos, Cell, 2012, 149, 358–370 CrossRef CAS PubMed.
- A. Ablasser, M. Goldeck, T. Cavlar, T. Deimling, G. Witte, I. Roehl, K.-P. Hopfner, J. Ludwig and V. Hornung, Nature, 2013, 498, 380–384 CrossRef CAS PubMed.
- X. Cai, Y. H. Chiu and Z. J. Chen, Mol. Cell, 2014, 54, 289–296 CrossRef CAS PubMed.
- T. S. Xiao and K. A. Fitzgerald, Mol. Cell, 2013, 51, 135–139 CrossRef CAS PubMed.
- R. M. Corrigan and A. Gründling, Nat. Rev. Microbiol., 2013, 11, 513–524 CrossRef CAS PubMed.
- G. B. Hecht and A. Newton, J. Bacteriol., 1995, 177, 6223–6229 CrossRef CAS PubMed.
- R. Tal, H. C. Wong, R. Calhoon, D. Gelfand, A. L. Fear, G. Volman, R. Mayer, P. Ross, D. Amikam, H. Weinhouse, A. Cohen, S. Sapir, P. Ohana and M. Benziman, J. Bacteriol., 1998, 180, 4416–4425 CAS.
- N. De, M. V. A. S. Navarro, R. V. Raghavan and H. Sondermann, J. Mol. Biol., 2009, 393, 619–633 CrossRef CAS PubMed.
- R. Simm, M. Morr, A. Kader, M. Nimtz and U. Römling, Mol. Microbiol., 2004, 53, 1123–1134 CrossRef CAS PubMed.
- M. Tarnawski, T. R. Barends and I. Schlichting, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2015, 71, 2158–2177 CrossRef CAS PubMed.
- J. L. Hunter, G. B. Severin, B. J. Koestler and C. M. Waters, BMC Microbiol., 2014, 14, 22 CrossRef PubMed.
- D. Pérez-Mendoza, S. J. Coulthurst, S. Humphris, E. Campbell, M. Welch, I. K. Toth and G. P. Salmond, Mol. Microbiol., 2011, 82, 719–733 CrossRef PubMed.
- N. De, M. Pirruccello, P. V. Krasteva, N. Bae, R. V. Raghavan and H. Sondermann, PLoS Biol., 2008, 6, 601–617 CrossRef CAS PubMed.
- C. Chan, R. Paul, D. Samoray, N. C. Amiot, B. Giese, U. Jenal and T. Schirmer, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17084–17089 CrossRef CAS PubMed.
- M. W. Chen, M. Kotaka, C. Vonrhein, G. Bricogne, F. Rao, M. L. C. Chuah, D. Svergun, G. Schneider, Z.-X. Liang and J. Lescar, J. Bacteriol., 2012, 194, 4837–4846 CrossRef CAS PubMed.
- F. Rao, Y. Yang, Y. Qi and Z. X. Liang, J. Bacteriol., 2008, 190, 3622–3631 CrossRef CAS PubMed.
- M. W. Orr, G. P. Donaldson, G. B. Severin, J. Wang, H. O. Sintim, C. M. Waters and V. T. Lee, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E5048–E5057 CrossRef CAS PubMed.
- D. Cohen, U. Mechold, H. Nevenzal, Y. Yarmiyhu, T. E. Randall, D. C. Bay, J. D. Rich, M. R. Parsek, V. Kaever, J. J. Harrison and E. Banin, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11359–11364 CrossRef CAS PubMed.
- S. Ghosh and M. P. Deutscher, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 4372–4377 CrossRef CAS.
- S. K. Niyogi and A. K. Datta, J. Biol. Chem., 1975, 250, 7307–7312 CAS.
- D. Yu and M. P. Deutscher, J. Bacteriol., 1995, 177, 4137–4139 CrossRef CAS PubMed.
- M. Y. Galperin, A. N. Nikolskaya and E. V. Koonin, FEMS Microbiol. Lett., 2001, 204, 213–214 CAS.
- R. P. Ryan, Y. Fouhy, J. F. Lucey, L. C. Crossman, S. Spiro, Y. W. He, L. H. Zhang, S. Heeb, M. Camara, P. Williams and J. M. Dow, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 6712–6717 CrossRef CAS PubMed.
- A. L. Lovering, M. J. Capeness, C. Lambert, L. Hobley and R. E. Sockett, mBio, 2011, 2, e00163 CrossRef PubMed.
- D. Bellini, D. L. Caly, Y. McCarthy, M. Bumann, S. Q. An, J. M. Dow, R. P. Ryan and M. A. Walsh, Mol. Microbiol., 2014, 91, 26–38 CrossRef CAS PubMed.
- S. Rinaldo, A. Paiardini, V. Stelitano, P. Brunotti, L. Cervoni, S. Fernicola, C. Protano, M. Vitali, F. Cutruzzolà and G. Giardina, J. Bacteriol., 2015, 197, 1525–1535 CrossRef CAS PubMed.
- C. D. Boyd and G. A. O'Toole, Annu. Rev. Cell Dev. Biol., 2012, 28, 439–462 CrossRef CAS PubMed.
- A. J. Wolfe and K. L. Visick, J. Bacteriol., 2008, 190, 463–475 CrossRef CAS PubMed.
- C. Lori, S. Ozaki, S. Steiner, R. Böhm, S. Abel, B. N. Dubey, T. Schirmer, S. Hiller and U. Jenal, Nature, 2015, 523, 236–239 CrossRef CAS PubMed.
- A. Duerig, S. Abel, M. Folcher, M. Nicollier, T. Schwede, N. Amiot, B. Giese and U. Jenal, Genes Dev., 2009, 23, 93–104 CrossRef CAS PubMed.
- R. Hengge, Nat. Rev. Microbiol., 2009, 7, 263–273 CrossRef CAS PubMed.
- Y. Kumagai, J. Matsuo, Y. Hayakawa and Y. Rikihisa, J. Bacteriol., 2010, 192, 4122–4133 CrossRef CAS PubMed.
- D. Amikam and M. Y. Galperin, Bioinformatics, 2006, 22, 3–6 CrossRef CAS PubMed.
- D. A. Ryjenkov, R. Simm, U. Römling and M. Gomelsky, J. Biol. Chem., 2006, 281, 30310–30314 CrossRef CAS PubMed.
- J. C. Whitney, G. B. Whitfield, L. S. Marmont, P. Yip, A. M. Neculai, Y. D. Lobsanov, H. Robinson, D. E. Ohman and P. L. Howell, J. Biol. Chem., 2015, 290, 12451–12462 CrossRef CAS PubMed.
- M. Christen, B. Christen, M. G. Allan, M. Folcher, P. Jenö, S. Grzesiek and U. Jenal, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 4112–4117 CrossRef CAS PubMed.
- J. T. Pratt, R. Tamayo, A. D. Tischler and A. Camilli, J. Biol. Chem., 2007, 282, 12860–12870 CrossRef CAS PubMed.
- C. Chan, R. Paul, D. Samoray, N. C. Amiot, B. Giese, U. Jenal and T. Schirmer, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17084–17089 CrossRef CAS PubMed.
- M. V. A. S. Navarro, N. De, N. Bae, Q. Wang and H. Sondermann, Structure, 2009, 17, 1104–1116 CrossRef CAS PubMed.
- S. Ozaki, A. Schalch-Moser, L. Zumthor, P. Manfredi, A. Ebbensgaard, T. Schirmer and U. Jenal, Mol. Microbiol., 2014, 94, 580–594 CrossRef CAS PubMed.
- T. Su, S. Liu, K. Wang, K. Chi, D. Zhu, T. Wei, Y. Huang, L. Guo, W. Hu, S. Xu, Z. Lin and L. Gu, J. Struct. Biol., 2015, 192, 1–13 CrossRef CAS PubMed.
- B. Y. Matsuyama, P. V. Krasteva, C. Baraquet, C. S. Harwood, H. Sondermann and M. V. Navarro, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E209–E218 CrossRef CAS PubMed.
- P. V. Krasteva, J. C. Fong, N. J. Shikuma, S. Beyhan, M. V. Navarro, F. H. Yildiz and H. Sondermann, Science, 2010, 327, 866–868 CrossRef CAS PubMed.
- N. Sudarsan, E. R. Lee, Z. Weinberg, R. H. Moy, J. N. Kim, K. H. Link and R. R. Breaker, Science, 2008, 321, 411–413 CrossRef CAS PubMed.
- E. R. Lee, J. L. Baker, Z. Weinberg, N. Sudarsan and R. R. Breaker, Science, 2010, 329, 845–848 CrossRef CAS PubMed.
- Z. Zhang, B. L. Gaffney and R. A. Jones, J. Am. Chem. Soc., 2004, 126, 16700–16701 CrossRef CAS PubMed.
- J. Wang, J. Zhou, G. P. Donaldson, S. Nakayama, L. Yan, Y. F. Lam, V. T. Lee and H. O. Sintim, J. Am. Chem. Soc., 2011, 133, 9320–9330 CrossRef CAS PubMed.
- J. Zhou, D. A. Sayre, J. Wang, N. Pahadi and H. O. Sintim, Molecules, 2012, 17, 13376–13389 CrossRef CAS PubMed.
- J. Zhao, E. Veliath, S. Kim, B. L. Gaffney and R. A. Jones, Nucleosides, Nucleotides Nucleic Acids, 2009, 28, 352–378 Search PubMed.
- K. Sambanthamoorthy, R. E. Sloup, V. Parashar, J. M. Smith, E. E. Kim, M. F. Semmelhack, M. B. Neiditch and C. M. Waters, Antimicrob. Agents Chemother., 2012, 56, 5202–5211 CrossRef CAS PubMed.
- S. Nakayama, J. Zhou, Y. Zheng, H. Szmacinski and H. O. Sintim, Future Sci. OA, 2016, 2 DOI:10.4155/fso.4115.4193.
- S. Nakayama, Y. Luo, J. Zhou, T. K. Dayie and H. O. Sintim, Chem. Commun., 2012, 48, 9059–9061 RSC.
- E. J. Diner, D. L. Burdette, S. C. Wilson, K. M. Monroe, C. A. Kellenberger, M. Hyodo, Y. Hayakawa, M. C. Hammond and R. E. Vance, Cell Rep., 2013, 3, 1355–1361 CrossRef CAS PubMed.
- C. A. Kellenberger, S. C. Wilson, J. Sales-Lee and M. C. Hammond, J. Am. Chem. Soc., 2013, 135, 4906–4909 CrossRef CAS PubMed.
- C. A. Kellenberger, C. Chen, A. T. Whiteley, D. A. Portnoy and M. C. Hammond, J. Am. Chem. Soc., 2015, 137, 6432–6435 CrossRef CAS PubMed.
- G. Tsuji and H. O. Sintim, Mol. BioSyst., 2016, 12, 773–777 RSC.
- S. Nakayama, I. Kelsey, J. Wang, K. Roelofs, B. Stefane, Y. Luo, V. T. Lee and H. O. Sintim, J. Am. Chem. Soc., 2011, 133, 4856–4864 CrossRef CAS PubMed.
- S. Nakayama, I. Kelsey, J. Wang and H. O. Sintim, Chem. Commun., 2011, 47, 4766–4768 RSC.
- B. T. Roembke, J. Zhou, Y. Zheng, D. Sayre, A. Lizardo, L. Bernard and H. O. Sintim, Mol. BioSyst., 2014, 10, 1568–1575 RSC.
- S. Fernicola, A. Paiardini, G. Giardina, G. Rampioni, L. Leoni, F. Cutruzzolà and S. Rinaldo, J. Bacteriol., 2016, 198, 147–156 CrossRef CAS PubMed.
- O. J. Lieberman, M. W. Orr, Y. Wang and V. T. Lee, ACS Chem. Biol., 2014, 9, 183–192 CrossRef CAS PubMed.
- M. Y. Galperin, A. N. Nikolskaya and E. V. Koonin, FEMS Microbiol. Lett., 2001, 203, 11–21 CrossRef CAS PubMed.
- A. Kader, R. Simm, U. Gerstel, M. Morr and U. Römling, Mol. Microbiol., 2006, 60, 602–616 CrossRef CAS PubMed.
- Y. Li, S. Heine, M. Entian, K. Sauer and N. Frankenberg-Dinkel, J. Bacteriol., 2013, 195, 3531–3542 CrossRef CAS PubMed.
- O. J. Lieberman, J. J. DeStefano and V. T. Lee, PLoS One, 2013, 8, e53689 CrossRef CAS PubMed.
- K. Sambanthamoorthy, R. E. Sloup, V. Parashar, J. M. Smith, E. E. Kim, M. F. Semmelhack, M. B. Neiditch and C. M. Waters, Antimicrob. Agents Chemother., 2012, 56, 5202–5211 CrossRef CAS PubMed.
- K. Sambanthamoorthy, C. Luo, N. Pattabiraman, X. Feng, B. Koestler, C. M. Waters and T. J. Palys, Biofouling, 2014, 30, 17–28 CrossRef CAS PubMed.
- D. Antoniani, E. Rossi, S. Rinaldo, P. Bocci, M. Lolicato, A. Paiardini, N. Raffaelli, F. Cutruzzolà and P. Landini, Appl. Microbiol. Biotechnol., 2013, 97, 7325–7336 CrossRef CAS PubMed.
- D. Antoniani, P. Bocci, A. Maciag, N. Raffaelli and P. Landini, Appl. Microbiol. Biotechnol., 2010, 85, 1095–1104 CrossRef CAS PubMed.
- C. A. Shanahan and S. A. Strobel, Org. Biomol. Chem., 2012, 10, 9113–9129 Search PubMed.
- J. Zhou, S. Watt, J. Wang, S. Nakayama, D. A. Sayre, Y. F. Lam, V. T. Lee and H. O. Sintim, Bioorg. Med. Chem., 2013, 21, 4396–4404 CrossRef CAS PubMed.
- P. Ohana, D. P. Delmer, R. W. Carlson, J. Glushka, P. Azadi, T. Bacic and M. Benziman, Plant Cell Physiol., 1998, 39, 144–152 CrossRef CAS PubMed.
- C. A. Shanahan, B. L. Gaffney, R. A. Jones and S. A. Strobel, Biochemistry, 2013, 52, 365–377 CrossRef CAS PubMed.
- J. Wang, J. Zhou, G. P. Donaldson, S. Nakayama, L. Yan, Y. F. Lam, V. T. Lee and H. O. Sintim, J. Am. Chem. Soc., 2011, 133, 9320–9330 CrossRef CAS PubMed.
- C. A. Shanahan, B. L. Gaffney, R. A. Jones and S. A. Strobel, J. Am. Chem. Soc., 2011, 133, 15578–15592 CrossRef CAS PubMed.
- K. D. Smith, S. V. Lipchock and S. A. Strobel, Biochemistry, 2012, 51, 425–432 CrossRef CAS PubMed.
- S. L. Kuchma, A. E. Ballok, J. H. Merritt, J. H. Hammond, W. Lu, J. D. Rabinowitz and G. A. O'Toole, J. Bacteriol., 2010, 192, 2950–2964 CrossRef CAS PubMed.
- J. W. Hickman, D. F. Tifrea and C. S. Harwood, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 14422–14427 CrossRef CAS PubMed.
- H. Kulesekara, V. Lee, A. Brencic, N. Liberati, J. Urbach, S. Miyata, D. G. Lee, A. N. Neely, M. Hyodo, Y. Hayakawa, F. M. Ausubel and S. Lory, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 2839–2844 CrossRef CAS PubMed.
- W. J. Sandborn, E. C. Van O, B. J. Zins, W. J. Tremaine, D. C. Mays and J. J. Lipsky, Gastroenterology, 1995, 109, 1808–1817 CrossRef CAS.
- G. K. Azad and R. S. Tomar, Mol. Biol. Rep., 2014, 41, 4865–4879 CrossRef CAS PubMed.
- S. Thangamani, W. Younis and M. N. Seleem, Sci. Rep., 2015, 5, 11596 CrossRef CAS PubMed.
- Y. Ishihara, M. Hyodo, Y. Hayakawa, T. Kamegaya, K. Yamada, A. Okamoto, T. Hasegawa and M. Ohta, FEMS Microbiol. Lett., 2009, 301, 193–200 CrossRef CAS PubMed.
- B. L. Gaffney and R. A. Jones, Org. Lett., 2014, 16, 158–161 CrossRef CAS PubMed.
- T. Fujino, K. Okada and H. Isobe, Tetrahedron Lett., 2014, 55, 2659–2661 CrossRef CAS.
- C. Kinzie, A. Steele, S. Pasciolla and W. Wuest, Tetrahedron Lett., 2014, 55, 4966–4968 CrossRef CAS.
- S. Fernicola, I. Torquati, A. Paiardini, G. Giardina, G. Rampioni, M. Messina, L. Leoni, F. Del Bello, R. Petrelli, S. Rinaldo, L. Cappellacci and F. Cutruzzolà, J. Med. Chem., 2015, 58, 8269–8284 CrossRef CAS PubMed.
- G. Witte, S. Hartung, K. Buettner and K.-P. Hopfner, Mol. Cell, 2008, 30, 167–178 CrossRef CAS PubMed.
- M. Bejerano-Sagie, Y. Oppenheimer-Shaanan, I. Berlatzky, A. Rouvinski, M. Meyerovich and S. Ben-Yehuda, Cell, 2006, 125, 679–690 CrossRef CAS PubMed.
- R. M. Corrigan, J. C. Abbott, H. Burhenne, V. Kaever and A. Gründling, PLoS Pathog., 2011, 7, e1002217 CrossRef CAS PubMed.
- J. J. Woodward, A. T. Iavarone and D. A. Portnoy, Science, 2010, 328, 1703–1705 CrossRef CAS PubMed.
- Y. Bai, J. Yang, X. Zhou, X. Ding, L. E. Eisele and G. Bai, PLoS One, 2012, 7, e35206 CrossRef CAS PubMed.
- T. Kamegaya, K. Kuroda and Y. Hayakawa, Nagoya J. Med. Sci., 2011, 73, 49–57 CAS.
- J. R. Barker, B. J. Koestler, V. K. Carpenter, D. L. Burdette, C. M. Waters, R. E. Vance and R. H. Valdivia, mBio, 2013, 4, e00018 CrossRef CAS PubMed.
- L. Zhang, W. Li and Z. G. He, J. Biol. Chem., 2013, 288, 3085–3096 CrossRef CAS PubMed.
- Y. Bai, J. Yang, T. M. Zarrella, Y. Zhang, D. W. Metzger and G. Bai, J. Bacteriol., 2014, 196, 614–623 CrossRef PubMed.
- C. E. Witte, A. T. Whiteley, T. P. Burke, J.-D. Sauer, D. A. Portnoy and J. J. Woodward, mBio, 2013, 4, e00282 CrossRef CAS PubMed.
- X. Peng, Y. Zhang, G. Bai, X. Zhou and H. Wu, Mol. Microbiol., 2016, 99, 945–959 CrossRef CAS PubMed.
- Y. Oppenheimer-Shaanan, E. Wexselblatt, J. Katzhendler, E. Yavin and S. Ben-Yehuda, EMBO Rep., 2011, 12, 594–601 CrossRef CAS PubMed.
- Q. Tang, Y. Luo, C. Zheng, K. Yin, M. K. Ali, X. Li and J. He, Int. J. Biol. Sci., 2015, 11, 813–824 CrossRef CAS PubMed.
- Y. Luo and J. D. Helmann, Mol. Microbiol., 2012, 83, 623–639 CrossRef CAS PubMed.
- M. Müller, T. Deimling, K. P. Hopfner and G. Witte, Biochem. J., 2015, 469, 367–374 CrossRef PubMed.
- F. M. Mehne, K. Schröder-Tittmann, R. T. Eijlander, C. Herzberg, L. Hewitt, V. Kaever, R. J. Lewis, O. P. Kuipers, K. Tittmann and J. Stülke, J. Biol. Chem., 2014, 289, 21098–21107 CrossRef CAS PubMed.
- T. Kamegaya, K. Kuroda and Y. Hayakawa, Nagoya J. Med. Sci., 2011, 73, 49–57 CAS.
- Y. Bai, J. Yang, L. E. Eisele, A. J. Underwood, B. J. Koestler, C. M. Waters, D. W. Metzger and G. Bai, J. Bacteriol., 2013, 195, 5123–5132 CrossRef CAS PubMed.
- J. Rosenberg, A. Dickmanns, P. Neumann, K. Gunka, J. Arens, V. Kaever, J. Stülke, R. Ficner and F. M. Commichau, J. Biol. Chem., 2015, 290, 6596–6606 CrossRef CAS PubMed.
- M. Müller, T. Deimling, K. P. Hopfner and G. Witte, Biochem. J., 2015, 469, 367–374 CrossRef PubMed.
- F. Rao, R. Y. See, D. Zhang, D. C. Toh, Q. Ji and Z.-X. Liang, J. Biol. Chem., 2010, 285, 473–482 CrossRef CAS PubMed.
- M. Ye, J.-J. Zhang, X. Fang, G. B. Lawlis, B. Troxell, Y. Zhou, M. Gomelsky, Y. Lou and X. F. Yang, Infect. Immun., 2014, 82, 1840–1849 CrossRef PubMed.
- K. Manikandan, V. Sabareesh, N. Singh, K. Saigal, U. Mechold and K. M. Sinha, PLoS One, 2014, 9, e86096 CrossRef PubMed.
- J. Yang, Y. Bai, Y. Zhang, V. D. Gabrielle, L. Jin and G. Bai, Mol. Microbiol., 2014, 93, 65–79 CrossRef CAS PubMed.
- T. N. Huynh, S. Luo, D. Pensinger, J. D. Sauer, L. Tong and J. J. Woodward, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E747–E756 CrossRef CAS PubMed.
- Q. He, F. Wang, S. Liu, D. Zhu, H. Cong, F. Gao, B. Li, H. Wang, Z. Lin, J. Liao and L. Gu, J. Biol. Chem., 2016, 291, 3668–3681 CrossRef CAS PubMed.
- C. Zheng, Y. Ma, X. Wang, Y. Xie, M. K. Ali and J. He, Front. Microbiol., 2015, 6, 908 Search PubMed.
- R. M. Corrigan, I. Campeotto, T. Jeganathan, K. G. Roelofs, V. T. Lee and A. Gründling, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9084–9089 CrossRef CAS PubMed.
- J. Gundlach, A. Dickmanns, K. Schröder-Tittmann, P. Neumann, J. Kaesler, J. Kampf, C. Herzberg, E. Hammer, F. Schwede, V. Kaever, K. Tittmann, J. Stülke and R. Ficner, J. Biol. Chem., 2015, 290, 3069–3080 CrossRef CAS PubMed.
- I. Campeotto, Y. Zhang, M. G. Mladenov, P. S. Freemont and A. Gründling, J. Biol. Chem., 2015, 290, 2888–2901 CrossRef CAS PubMed.
- K. Sureka, P. H. Choi, M. Precit, M. Delince, D. A. Pensinger, T. N. Huynh, A. R. Jurado, Y. A. Goo, M. Sadilek, A. T. Iavarone, J. D. Sauer, L. Tong and J. J. Woodward, Cell, 2014, 158, 1389–1401 CrossRef CAS PubMed.
- J. W. Nelson, N. Sudarsan, K. Furukawa, Z. Weinberg, J. X. Wang and R. R. Breaker, Nat. Chem. Biol., 2013, 9, 834–839 CrossRef CAS PubMed.
- P. Y. Watson and M. J. Fedor, Nat. Chem. Biol., 2012, 8, 963–965 CrossRef CAS PubMed.
- A. J. Underwood, Y. Zhang, D. W. Metzger and G. Bai, J. Microbiol. Methods, 2014, 107, 58–62 CrossRef CAS PubMed.
- J. Zhou, D. A. Sayre, Y. Zheng, H. Szmacinski and H. O. Sintim, Anal. Chem., 2014, 86, 2412–2420 CrossRef CAS PubMed.
- Y. Zheng, J. Zhou, D. A. Sayre and H. O. Sintim, Chem. Commun., 2014, 50, 11234–11237 RSC.
- C. Opoku-Temeng and H. O. Sintim, Chem. Commun., 2016, 52, 3754–3757 RSC.
- C. Opoku-Temeng and H. O. Sintim, Sci. Rep., 2016, 6, 25445, DOI:10.1038/srep25445.
- R. E. Meehan, C. D. Torgerson, B. L. Gaffney, R. A. Jones and S. A. Strobel, Biochemistry, 2016, 55, 837–849 CrossRef CAS PubMed.
- Y. Zheng, J. Zhou, D. A. Sayre and H. O. Sintim, Chem. Commun., 2014, 50, 11234–11237 RSC.
- J. Gao, J. Tao, W. Liang, M. Zhao, X. Du, S. Cui, H. Duan, B. Kan, X. Su and Z. Jiang, Cell Res., 2015, 25, 539–550 CrossRef CAS PubMed.
- C. A. Shanahan, B. L. Gaffney, R. A. Jones and S. A. Strobel, J. Am. Chem. Soc., 2011, 133, 15578–15592 CrossRef CAS PubMed.
- C. A. Kellenberger, S. C. Wilson, S. F. Hickey, T. L. Gonzalez, Y. Su, Z. F. Hallberg, T. F. Brewer, A. T. Iavarone, H. K. Carlson, Y. F. Hsieh and M. C. Hammond, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5383–5388 CrossRef CAS PubMed.
- J. W. Nelson, N. Sudarsan, G. E. Phillips, S. Stav, C. E. Lünse, P. J. McCown and R. R. Breaker, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5389–5394 CrossRef CAS PubMed.
- X. Zhang, H. Shi, J. Wu, X. Zhang, L. Sun, C. Chen and Z. J. Chen, Mol. Cell, 2013, 51, 226–235 CrossRef CAS PubMed.
- P. Gao, M. Ascano, Y. Wu, W. Barchet, B. L. Gaffney, T. Zillinger, A. A. Serganov, Y. Liu, R. A. Jones, G. Hartmann, T. Tuschl and D. J. Patel, Cell, 2013, 153, 1094–1107 CrossRef CAS PubMed.
- J. Wu, L. Sun, X. Chen, F. Du, H. Shi, C. Chen and Z. J. Chen, Science, 2013, 339, 826–830 CrossRef CAS PubMed.
- L. Sun, J. Wu, F. Du, X. Chen and Z. J. Chen, Science, 2013, 339, 786–791 CrossRef CAS PubMed.
- L. Li, Q. Yin, P. Kuss, Z. Maliga, J. L. Millán, H. Wu and T. J. Mitchison, Nat. Chem. Biol., 2014, 10, 1043–1048 CrossRef CAS PubMed.
- K. Kato, H. Nishimasu, S. Okudaira, E. Mihara, R. Ishitani, J. Takagi, J. Aoki and O. Nureki, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16876–16881 CrossRef CAS PubMed.
- X. Zhang, J. Wu, F. Du, H. Xu, L. Sun, Z. Chen, C. A. Brautigam and Z. J. Chen, Cell Rep., 2014, 6, 421–430 CrossRef CAS PubMed.
- V. Hornung and E. Latz, Nat. Rev. Immunol., 2010, 10, 123–130 CrossRef CAS PubMed.
- Y. Tanaka and Z. J. Chen, Sci. Signaling, 2012, 5, ra20 CrossRef PubMed.
- T. Abe, A. Harashima, T. Xia, H. Konno, K. Konno, A. Morales, J. Ahn, D. Gutman and G. N. Barber, Mol. Cell, 2013, 50, 5–15 CrossRef CAS PubMed.
- D. L. Burdette, K. M. Monroe, K. Sotelo-Troha, J. S. Iwig, B. Eckert, M. Hyodo, Y. Hayakawa and R. E. Vance, Nature, 2011, 478, 515–518 CrossRef CAS PubMed.
- D. K. Karaolis, M. H. Rashid, R. Chythanya, W. Luo, M. Hyodo and Y. Hayakawa, Antimicrob. Agents Chemother., 2005, 49, 1029–1038 CrossRef CAS PubMed.
- D. K. R. Karaolis, T. K. Means, D. Yang, M. Takahashi, T. Yoshimura, E. Muraille, D. Philpott, J. T. Schroeder, M. Hyodo, Y. Hayakawa, B. G. Talbot, E. Brouillette and F. Malouin, J. Immunol., 2007, 178, 2171–2181 CrossRef CAS.
- D. K. R. Karaolis, K. R. Cheng, M. Lipsky, A. Elnabawi, J. Catalano, M. Hyodo, Y. Hayakawa and J. P. Raufman, Biochem. Biophys. Res. Commun., 2005, 329, 40–45 CrossRef CAS PubMed.
- D. Chandra, W. Quispe-Tintaya, A. Jahangir, D. Asafu-Adjei, I. Ramos, H. O. Sintim, J. Zhou, Y. Hayakawa, D. K. Karaolis and C. Gravekamp, Cancer Immunol. Res., 2014, 2, 901–910 CrossRef CAS PubMed.
- S. Yildiz, E. Alpdundar, B. Gungor, T. Kahraman, B. Bayyurt, I. Gursel and M. Gursel, Eur. J. Immunol., 2015, 45, 1170–1179 CrossRef CAS PubMed.
- B. J. Koestler, S. S. Seregin, D. P. Rastall, Y. A. Aldhamen, S. Godbehere, A. Amalfitano and C. M. Waters, Clin. Vaccine Immunol., 2014, 21, 1550–1559 CrossRef PubMed.
- F. S. Alyaqoub, Y. A. Aldhamen, B. J. Koestler, E. L. Bruger, S. S. Seregin, C. Pereira-Hicks, S. Godbehere, C. M. Waters and A. Amalfitano, J. Immunol., 2016, 196, 1741–1752 CrossRef CAS PubMed.
- H. Miyabe, M. Hyodo, T. Nakamura, Y. Sato, Y. Hayakawa and H. Harashima, J. Controlled Release, 2014, 184, 20–27 CrossRef CAS PubMed.
- T. Nakamura, H. Miyabe, M. Hyodo, Y. Sato, Y. Hayakawa and H. Harashima, J. Controlled Release, 2015, 216, 149–157 CrossRef CAS PubMed.
- X. D. Li, J. Wu, D. Gao, H. Wang, L. Sun and Z. J. Chen, Science, 2013, 341, 1390–1394 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |