
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A metal sulfide photocatalyst composed of ubiquitous elements for solar hydrogen production†
Y.
Shiga
a,
N.
Umezawa
*b,
N.
Srinivasan
a,
S.
Koyasu
a,
E.
Sakai
a and
M.
Miyauchi
*a
aSchool of Materials and Chemical Technology, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro, Tokyo 152-8552, Japan. E-mail: mmiyauchi@ceram.titech.ac.jp
bInternational Center for Materials Nanoarchitectonics, National Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan. E-mail: Umezawa.Naoto@nims.go.jp
First published on 12th May 2016
Abstract
A visible-light-sensitive tin sulfide photocatalyst was designed based on a ubiquitous element strategy and density functional theory (DFT) calculations. Computational analysis suggested that tin monosulfide (SnS) would be more efficient than SnS2 as a photocathode for hydrogen production because of the low ionization potential and weak ionic character of SnS. To test this experimentally, nanoparticles of SnS were loaded onto a mesoporous electrode using a wet chemical method, and the bandgap of the synthesized SnS quantum dots was found to be tunable by adjusting the number of successive ionic layer adsorption and reaction (SILAR) cycles, which controls the magnitude of the quantum confinement effect. Efficient hydrogen production was achieved when the bandgap of SnS was wider than that of the bulk form.
For efficient photocatalytic solar water splitting, materials with visible-light harvesting ability and electrons and/or holes with high mobilities for promoting charge separation are required. Several efficient metal oxide semiconductor photocatalysts, which typically have d0 or d10 metal orbital configurations, have been reported to date.1–3 Most of these metal oxides have a wide bandgap, as their valence band consists of a deep oxygen 2p orbital, and hole carriers have a relatively heavy effective mass due to the localized nature of the oxygen 2p state. In contrast to metal oxides, metal sulfides generally have a shallow valence band4 and exhibit a clear quantum size effect due to their small effective mass.5 These properties contribute to the visible-light harvesting ability and high charge mobility of metal sulfides.6,7 Several previous studies have examined the photo-energy conversion of metal sulfide quantum dots (QDs), including CdS, CuInS2, and PbS QDs,8–12 as the bandgap, and conduction and valence band potentials of the materials are tunable based on their particle size.13,14 However, because these metal sulfides are composed of toxic and/or rare elements, the design of metal sulfide semiconductors composed of safe and abundant elements is required for use in practical applications. This approach is consistent with the “ubiquitous element strategy” that has recently been proposed for materials used in industrial activities and has attracted considerable attention towards a safe and sustainable society.15–17 Previous studies reported the efficient materials for solar-to-energy conversion with ubiquitous earth-abundant elements, like carbon nitride and boron nitride.18–20
Among the various elements in the periodic table, we have focused on group IV–VI chalcogenides due to their excellent optoelectronic properties and high charge mobilities.21 In particular, tin sulfides, which have unique electrical characteristics, various valence numbers, and sensitivity to a wide range of visible light, are attractive materials for use in electronic semiconductor devices and thin-film solar cells.22,23 Tin sulfides have two chemical compositions: tin monosulfide (SnS) and tin disulfide (SnS2). Herein, the electronic structures of SnS and SnS2 were elucidated based on density functional theory (DFT) calculations, and the semiconductor characteristics of these compounds were also evaluated. In addition, SnS and SnS2 QDs were synthesized by a facile wet chemical method. For the synthesis, a mesoporous TiO2 electrode was used as the support, since minute pores are helpful for fixing QDs on the electrode support. The deposition of nanometer-scale metal sulfide QDs is expected to induce the quantum confinement effect and improve the photochemical properties of the material compared to the bulk form. The photoelectrochemical properties and photocatalytic hydrogen production activities of the tin sulfide QD-modified electrodes were also evaluated. Further, the effect of a thin layer of amorphous SiO2 on the stability of the tin sulfide QDs was examined under visible-light irradiation.
Fig. 1 shows the calculated band diagrams of SnS and SnS2. The bandgap of SnS was narrower than that of SnS2, and both the ionization potential and electron affinity of SnS were smaller than those of SnS2. The conduction band potential of SnS is higher than the potential for hydrogen production. Band dispersion and density of states were also calculated for SnS and SnS2 and showed that both materials are indirect transition semiconductors, as shown in the ESI† (Fig. S1), which was consistent with the previous reports.24–28 Notably, the valence band of SnS consisted of Sn-5s and S-3p hybrid orbitals, suggesting that SnS would function as a p-type semiconductor. Burton et al.22,23 also reported a similar band structure for tin sulfides and suggested that SnS would be a suitable semiconductor for thin-film solar cells. Based on our DFT calculations, SnS was also predicted to be potentially useful as a photocathode for hydrogen production, although SnS is unable to evolve oxygen from water. In contrast, SnS2 would not produce hydrogen from water because of its low conduction band potential.
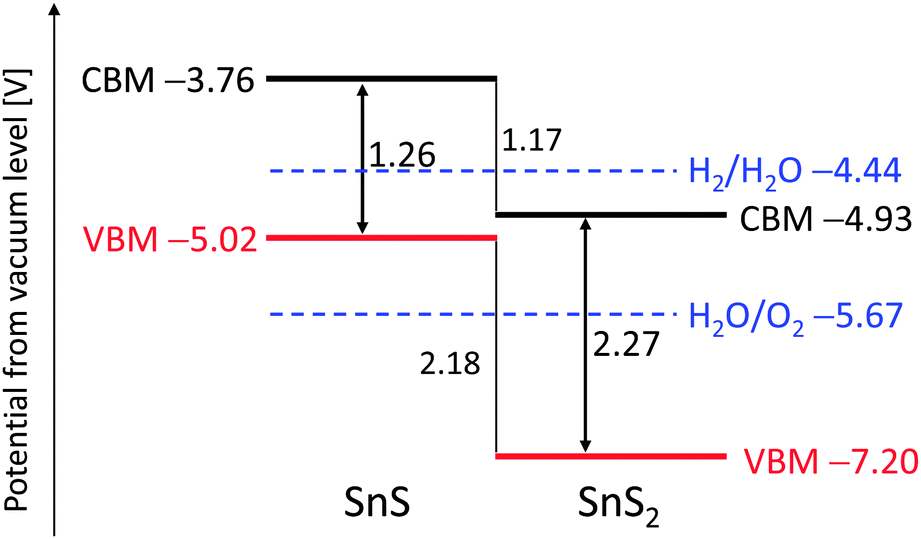 | ||
| Fig. 1 Band diagram of SnS and SnS2 based on DFT calculations. | ||
We loaded SnS QDs onto mesoporous TiO2 electrodes using the successive ionic layer adsorption and reaction (SILAR) method,29,30 and SnS2 particles were loaded onto TiO2 electrodes using a hydrothermal reaction.31 Detailed experimental procedures are described in the ESI.† The crystal phases of SnS and SnS2 were Herzenbergite (ICDD No. 00-039-0354) and Berndtite (ICDD No. 00-023-0677), respectively, as determined by X-ray diffraction patterns (XRD, Fig. 2a). Analysis of X-ray photoelectron spectroscopy (XPS) spectra for the Sn-3d and S-2p orbitals (Fig. 2b and c) revealed the chemical shift, owing to differences in the valence numbers of SnS and SnS2.32 In addition, chemical composition analysis by inductively coupled plasma (ICP) spectroscopy revealed that the atomic ratios of S/Sn in SnS and SnS2 were nearly 1 and 2, respectively. The particle sizes of SnS (10 SILAR cycles) and SnS2 (150 °C hydrothermal temperature) were determined to be approximately 10 nm, based on TEM observation (Fig. S2, ESI†).
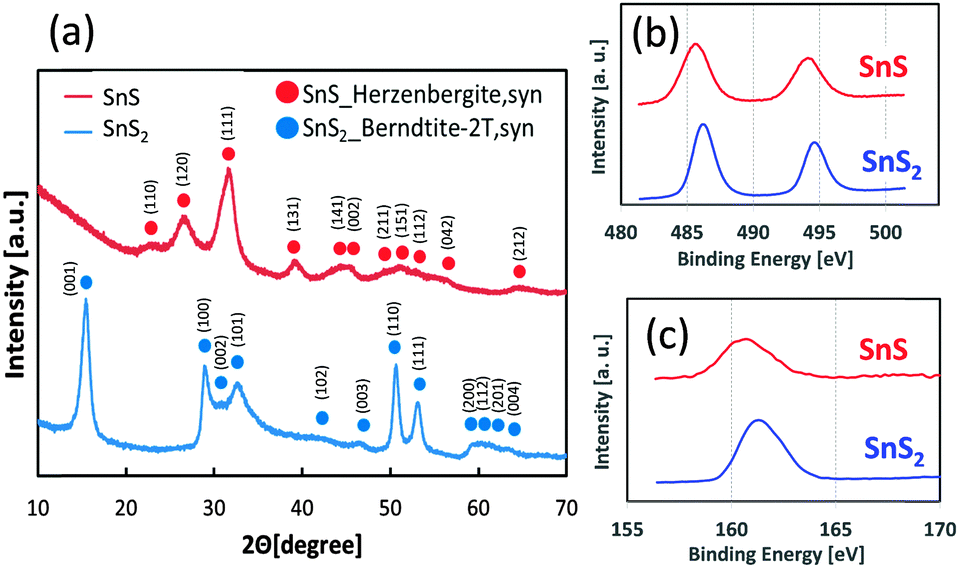 | ||
| Fig. 2 XRD diffraction patterns of SnS and SnS2 powder (a), and XPS spectra for Sn-3d (b) and S-2p orbitals (c). | ||
The optical absorption properties of SnS and SnS2 are shown in Fig. 3a. The bandgap of SnS appeared to be narrower than that of SnS2, a finding that is consistent with the DFT calculations. Notably, the bandgap of SnS was dependent on the number of SILAR cycles. Specifically, with increasing number of SILAR cycles, the absorption of the SnS particles shifted towards lower energy, a property that was attributed to the quantum confinement effect. The colour of SnS changed from orange to brown with the increasing number of SILAR cycles, whereas the SnS2 electrode was yellow in colour (Fig. S4, ESI†).
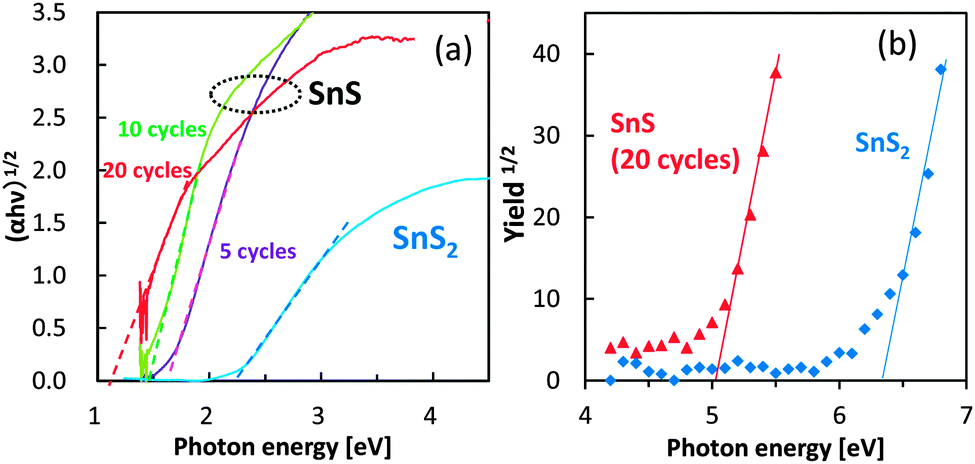 | ||
| Fig. 3 UV-Vis spectra (a) and ultraviolet photoelectron spectra (b) of SnS electrodes. The SnS electrodes in panel (a) were prepared using the SILAR method with 5, 10, and 20 cycles. | ||
The ultraviolet photoelectron spectra of SnS and SnS2 were next determined to examine the valence band potential of these sulfides (Fig. 3b). The ionization potential of SnS was lesser than that of SnS2, suggesting that SnS had a shallower valence band. The observed valence band positions were also consistent with the DFT calculations.
The relationship between photocurrent densities and bandgap values for the SnS and SnS2 electrodes was also investigated under chopped irradiation by a solar simulator without bias application (Fig. 4a). A small photocurrent was detected from the bare TiO2 electrode used as the baseline control (Fig. 4a, dashed line) and was attributed to the small amount of UV light that passed through the optical UV cut-off filter. Photocurrent generation by SnS2 was similar to that of bare TiO2, suggesting that the hydrogen generation rate in the SnS2 system was limited. In contrast, photocurrent by the SnS electrodes was significant, and the optimum bandgap value for the maximal photocurrent was approximately 1.4 eV, which was wider than the bulk bandgap value. Photon energy with 1.4 eV matches with the solar light spectrum. If the bandgap is wider than 1.5 eV, the number of electron–hole pairs are reduced due to the limited optical absorption. On the other hand, as the bandgap narrows with increasing number of SILAR cycles, the reduction potential of SnS was limited for hydrogen production. Further, as the bandgap narrows with increasing number of SILAR cycles, the TiO2 electrode surface becomes overloaded with SnS as free particles, which do not have contact with the mesoporous support. Under this condition, electron transport to the Pt electrode is limited. Also, overloaded SnS particles prevent the diffusion of electrolyte agent to the active SnS particles attached on TiO2. Therefore, the bandgap value of 1.4 eV for SnS determined in the present study appears to be optimal for solar hydrogen production. The results of control experiments, such as that using the SnS/TiO2 electrode without the Pt layer, are shown in our ESI† (Fig. S6), but the hydrogen production from these electrodes was negligible.
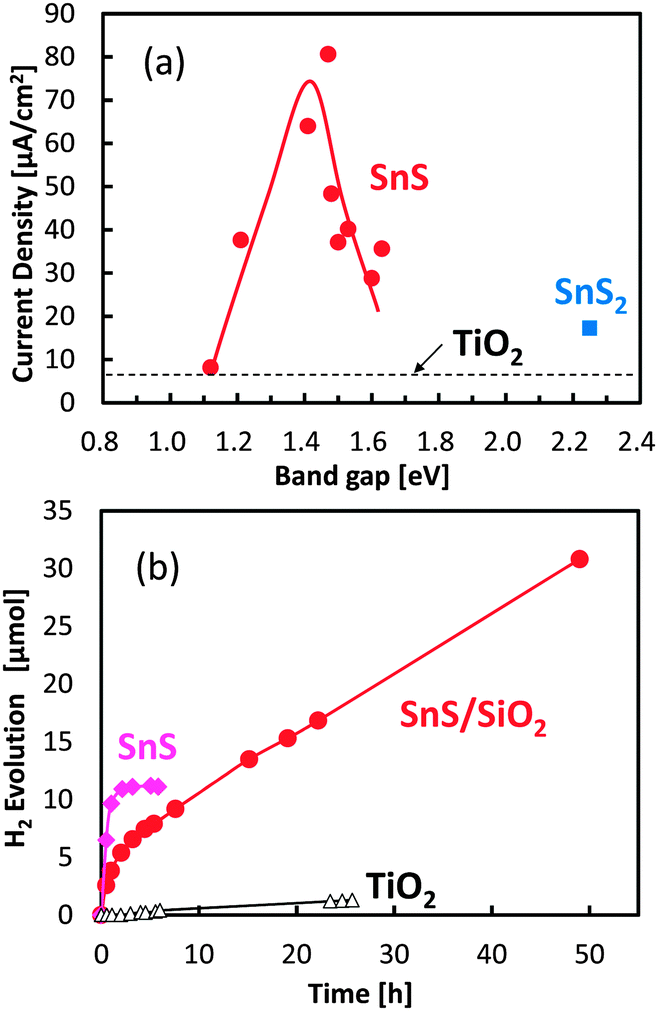 | ||
| Fig. 4 Photocurrent density under an AM 1.5 M solar simulator versus the bandgap energy of the semiconductor (a), and photocatalytic hydrogen evolution under visible-light irradiation (b). In both (a) and (b), visible light was irradiated through a UV cut-off filter, and a bias potential was not applied on the electrode. For the measurement of photocurrent densities, the Pt wire and the Ag/AgCl electrode were used as counter and reference electrodes, respectively. For the analysis of hydrogen production, a thin Pt layer was inserted between FTO and the mesoporous TiO2 layer using a sputtering method as a cocatalyst for hydrogen production. The electrolyte solution contained Na2S and Na2SO3 as sacrificial agents for the oxidation reaction. | ||
Photocatalytic hydrogen production by SnS with an optimized bandgap was further examined under visible-light irradiation without the application of a bias potential (Fig. 4b). SnS produced hydrogen under photon flux, and the quantum efficiency was calculated to be 0.31%, which was determined by the number of reacted electrons versus the absorbed photon numbers (see our ESI†). However, hydrogen production ceased after several hours of irradiation, owing to the photo-corrosion of SnS. In an attempt to avoid the photo-corrosion of SnS, a thin layer of amorphous silica (SiO2) with a thickness of less than 2 nm (Fig. S7, ESI†) was coated on the SnS electrode. Although the initial hydrogen production rate of the SiO2-coated SnS electrode was lower than that of the uncoated electrode, the stability of the SnS photocatalyst was significantly improved by the thin SiO2 coating. The calculated turnover number for the SiO2-coated SnS electrode was 4.2 after 50 h of visible-light irradiation, and hydrogen production continued to be produced even after 50 h. The improved stability is attributed to passivation by the aqueous electrolyte and/or tunnelling of charge carriers through the nanometer-scale SiO2 layer.33 The brown colour of the SnS electrode without the SiO2 layer became white under visible light irradiation, while its color was relatively retained by SiO2 coating.
We also evaluated the oxygen production properties of SnS and SnS2 in the presence of aqueous Ag+ as a reductive sacrificial agent; however, neither compound exhibited photocatalytic activity for water oxidation under these conditions. The valence band of SnS is not deep enough to produce oxygen, and SnS2 was corroded by visible light-induced oxidation. Based on these results, we can conclude that SnS is a promising candidate for a visible-light sensitive photocathode for hydrogen production and can be combined with a photoanode to construct a Z-scheme system for efficient overall water splitting.
In conclusion, density functional theory calculations were used to determine the electronic structure of the tin sulfides SnS and SnS2 and suggested that SnS is a potentially suitable photocathode for solar hydrogen production. Specifically, SnS has sufficiently low electron affinity to produce hydrogen from water and also has a small ionization potential, allowing for the absorption of a wide range of visible light. We experimentally synthesized SnS QDs using the SILAR method and demonstrated that this material could be used for efficient photocatalytic hydrogen production when the bandgap of SnS was wider than that of the bulk material. Because SnS is composed of safe and abundant elements, and SnS QDs can be synthesized using a facile wet chemical method that is applicable for mass production, we anticipate that the SnS QDs described here can be applied as photocathodes for solar hydrogen production.
We thank Mr K. Hori and Mr J. Koki at the Center for Advanced Materials Analysis of the Tokyo Institute of Technology for performing the TEM and SEM observations. We also appreciate the help of RIKEN KEIKI Co., Ltd for the UPS measurements and Ms G. Yin at the Tokyo Institute of Technology for the XPS analysis. We acknowledge Mr G. Newton for the critical reading of the manuscript. This research was supported by JST-PRESTO and JST-ACT-C programs, and JSPS KAKENHI Grant Number 26410234.
Notes and references
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295 CrossRef CAS PubMed.
- Q. Wang, T. Hisatomi, Q. Jia, H. Tokudome, M. Zhong, C. Wang, Z. Pan, T. Takata, M. Nakabayashi, N. Shibata, Y. Li, I. D. Sharp, A. Kudo, T. Yamada and K. Domen, Nat. Mater., 2016 DOI:10.1038/nmat4589.
- S. Narushima, M. Hiroki, K. Ueda, K.-I. Shimizu, T. Kamiya, M. Hirano and H. Hosono, Philos. Mag. Lett., 2004, 84, 665–671 CrossRef CAS.
- A. J. Hoffman, G. Mills, H. Yee and M. R. Hoffmann, J. Phys. Chem., 1992, 96, 5546–5552 CrossRef CAS.
- I. Tsuji, H. Kato and A. Kudo, Angew. Chem., 2005, 117, 3631–3634 CrossRef.
- W. Septina, Gunawan, S. Ikeda, T. Harada, M. Higashi, R. Abe and M. Matsumura, J. Phys. Chem. C, 2015, 119, 8576–8583 CAS.
- D. R. Baker and P. V. Kamat, Adv. Funct. Mater., 2009, 19, 805–811 CrossRef CAS.
- R. Vogel, P. Hoyer and H. Weller, J. Phys. Chem., 1994, 98, 3183–3188 CrossRef CAS.
- Y. Jin-nouchi, T. Hattori, Y. Sumida, M. Fujishima and H. Tada, ChemPhysChem, 2010, 11, 3592–3595 CrossRef CAS PubMed.
- Q. Shen, D. Arae and T. Toyoda, J. Photochem. Photobiol., A, 2004, 164, 75–80 CrossRef CAS.
- J. Y. Chang, J. M. Lin, L. F. Su and C. F. Chang, ACS Appl. Mater. Interfaces, 2013, 5, 8740–8752 CAS.
- N. Srinivasan, Y. Shiga, D. Atarashi, E. Sakai and M. Miyauchi, Appl. Catal., B, 2015, 179, 113–121 CrossRef CAS.
- A. Kondo, G. Yin, N. Srinivasan, D. Atarashi, E. Sakai and M. Miyauchi, Nanoscale, 2015, 7, 12510–12515 RSC.
- H. Hosono, K. Hayashi, T. Kamiya, T. Atou and T. Susaki, Sci. Technol. Adv. Mater., 2011, 12, 034303 CrossRef.
- H. Takahashi, K. Igawa, K. Arii, Y. Kamihara, M. Hirano and H. Hosono, Nature, 2008, 453, 376–378 CrossRef CAS PubMed.
- Y. Hirayama, Y. K. Takahashi, S. Hirosawa and K. Hono, Scr. Mater., 2015, 95, 70–72 CrossRef CAS.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- C. Huang, C. Chen, M. Zhang, L. Lin, X. Ye, S. Lin, M. Antonietti and X. Wang, Nat. Commun., 2015, 6, 7698 CrossRef PubMed.
- J. Zhang, Y. Chen and X. Wang, Energy Environ. Sci., 2015, 8, 3092–3108 CAS.
- Y. Ogo, H. Hiramatsu, K. Nomura, H. Yanagi, T. Kamiya, M. Hirano and H. Hosono, Appl. Phys. Lett., 2008, 93, 032113 CrossRef.
- L. A. Burton, D. Colombara, R. D. Abellon, F. C. Grozema, L. M. Peter, T. J. Savenije, G. Dennler and A. Walsh, Chem. Mater., 2013, 25, 4908–4916 CrossRef CAS.
- L. A. Burton and A. Walsh, Appl. Phys. Lett., 2013, 102, 132111 CrossRef.
- W. Albers, C. Haas, H. J. Vink and J. D. Wasscher, J. Appl. Phys., 1961, 32, 2220–2225 CrossRef CAS.
- Z. Zainal, M. Z. Hussein and A. Ghazali, Sol. Energy Mater. Sol. Cells, 1996, 40, 347–357 CrossRef CAS.
- A. Sanchez-Juarez and A. Ortíz, Semicond. Sci. Technol., 2002, 17, 931 CrossRef CAS.
- L. Amalraj, C. Sanjeeviraja and M. Jayachandran, J. Cryst. Growth, 2002, 234, 683–689 CrossRef CAS.
- G. Domingo, R. S. Itoga and C. R. Kannewurf, Phys. Rev., 1966, 143, 536–541 CrossRef CAS.
- M. Miyauchi, Chem. Phys. Lett., 2011, 514, 151–155 CrossRef CAS.
- M. Miyauchi, Y. Shiga, N. Srinivasan, D. Atarashi and E. Sakai, Mater. Chem. Phys., 2015, 160, 383–388 CrossRef CAS.
- Y. C. Zhang, J. Li, M. Zhang and D. D. Dionysiou, Environ. Sci. Technol., 2011, 45, 9324–9331 CrossRef CAS PubMed.
- D. Chen, G. Shen, K. Tang, S. Lei, H. Zheng and Y. Qian, J. Cryst. Growth, 2004, 260, 469–474 CrossRef CAS.
- K. Zhao, Z. Pan, I. Mora-Seró, E. Cánovas, H. Wang, Y. Song, X. Gong, J. Wang, M. Bonn, J. Bisquert and X. Zhong, J. Am. Chem. Soc., 2015, 137, 5602–5609 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc03199d |
| This journal is © The Royal Society of Chemistry 2016 |