
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pharmaceutical crystallization with nanocellulose organogels†
Celia
Ruiz-Palomero
a,
Stuart R.
Kennedy
b,
M. Laura
Soriano
a,
Christopher D.
Jones
b,
Miguel
Valcárcel
*a and
Jonathan W.
Steed
*b
aDepartment of Analytical Chemistry, University of Córdoba, Marie Curie Building, Campus de Rabanales, E-14071 Córdoba, Spain. E-mail: qa1vacam@uco.es; Tel: +34 957 218616
bDepartment of Chemistry, Durham University, University Science Laboratories, South Road, Durham, DH1 3LE, UK. E-mail: jon.steed@durham.ac.uk; Fax: +44 (0)191 384 4737; Tel: +44 (0)191 334 2085
First published on 6th May 2016
Abstract
Carboxylated nanocellulose forms organogels at 0.3 wt% in the presence of a cationic surfactant. The resulting gels can be used as novel crystallization media for pharmaceutical solid form control, resulting in isolation a new sulfapyridine solvate, morphology modification and crystallization of an octadecylammonium salt of sulfamethoxazole.
The solid form of active pharmaceutical ingredients (APIs) continues to be of tremendous intellectual and commercial interest.1–5 The solid forms of a drug, which can be crystalline, co-crystalline, solvate or amorphous,6–9 can have different solubility, bioavailability, stability and processing characteristics.1,10 They also offer drug manufacturers new intellectual property opportunities.3,11,12 As a result the solid state chemistry of drug substances is far more intensively studied than most other molecular compounds. Generally new drug candidates undergo one or more rounds of solid form screening. While solid form screens are routine and can be undertaken relatively quickly, the reports of late-appearing, insoluble and hence more thermodynamically stable polymorphs of drugs such as ritonavir13 means that there is enduring interest in novel crystallization methods. Crystallization of a drug substance from common solvents may well readily reveal a variety of solid forms1 but crystallization strategies that can expand the solid form screening space beyond conventional methodology14 offers the opportunity to reveal hard-to-nucleate or slow-growing crystal forms. The issue is particularly topical since many computational crystal structure calculation approaches predict far more feasible polymorphs than are actually observed in practice.15,16 High-throughput approaches offer a greater chance of finding less-accessible polymorphs.17 Other novel approaches to solid form discovery include crystallization in microemulsion droplets,18,19 polymer microgels,20 under nanoscale confinement21 and epitaxial overgrowth on a related parent phase.22 In addition, recent work by ourselves and others23–26 has begun to develop a variation on the classical technique of crystallization from polymer hydrogels such as silica or agarose. By using small-molecule supramolecular gels this work has shown that gel phase crystallization can move from water to organic solvents and the gelation process can be made to be reversible under mild conditions through the use of competing supramolecular interactions such as anion binding.23,27–29 The potential advantages of this supramolecular organogel crystallization approach include diversity in solvent choice, the elimination of convection and the possibility of directed crystallization based on the local periodicity of the gel fibres.30 The controlled reversibility of the gelation process potentially allows for facile recovery of the gel-grown samples.
Nanocellulose (NC) exhibits a range of interesting properties such as a highly functionalised surface, a filamentous structure lending itself to gelation behaviour, high porosity, low coefficient of thermal expansion, optical transparency and interesting self-assembly behaviour. It is also an abundant and cheap material which has led to tremendous interest in the development of NC composites.31–34 Functionalised NC has promising properties as an active absorbent in analytical applications.35 In the present work we report the adaptation of functionalised nanocellulose for application as an organogel pharmaceutical crystallization medium.
Carboxylated nanocellulose (c-NC) can be prepared from microcrystalline cellulose by radical oxidation using the 2,2,6,6-tetramethylpiperidine-1-oxyl radical (TEMPO) and NaOCl.36 The oxidation process serves to disrupt the extensive inter-strand hydrogen bonding in microcrystalline cellulose and give separated nanocellulose filaments. The resulting c-NC forms an opaque hydrogel at around 9% by weight (see ESI,† Fig. S1) but is insoluble in most common organic solvents.35 For use as a pharmaceutical crystallization medium for relatively hydrophobic drugs with low water solubility we sought a simple strategy to render the resulting c-NC filaments more hydrophobic. We reasoned that pairing the surface carboxylic acid functionalities with organic primary ammonium cations should sheath the c-NC filaments in a hydrophobic coating rendering them soluble in organic solvents while also serving to isolate the filaments from one another and hence promote gelation properties. Gelation tests of c-NC in the presence of lipophilic amines were carried out in a range of organic solvents (toluene, chloroform, ethyl acetate, acetone, dimethyl sulfoxide (DMSO), dimethylformamide, ethanol, tetrahydrofuran, ethylene glycol, picoline, pyridine, nitrobenzene, dichlorobenzene and nitromethane). The following amines were used as c-NC solubilizers: undecylamine, dodecylamine, nonylamine, octadecylamine (OD), octylamine, amylamine, cyclopentylamine, tetrahydrofurfurylamine and 3-dimethylamino-1-propylamine. In general the majority of these experiments did not result in solubilisation of the c-NC. However the combination of the longest chain amine, octadecylamine, with c-NC in DMSO resulted in the formation of a robust, translucent gel. The gel was produced by vortexing and heating a solution of c-NC in the presence of amine in conjunction with brief (10 s) sonication37 in ultrasound bath. Gelation occurred on cooling to room temperature under ambient conditions. Optimisation of the gel revealed a critical gelation concentration of 0.3 wt% with 3 mg c-NC and 5 mg octadecylamine in 1 mL DMSO (Fig. 1A). The gel is thermoreversible with a Tgel of 58 °C (dropping ball method). The gel–sol transition is also observed as an endothermic transition by DSC with an onset temperature of 50 °C and a peak at 55 °C (ESI,† Fig. S2).
 | ||
| Fig. 1 (A) Optimised octadecylamine/c-NC gel in DMSO at 0.3 wt%. (B) SEM image of the OD/c-NC xerogel showing the layered structure. (C) Stress sweep rheometry of the OD/c-NC DMSO gel. | ||
The new OD/c-NC gel was dried under vacuum for 2 days, coated with 3 nm chromium and imaged by SEM which revealed an unusual structure involving an entangled sheet-like morphology, (Fig. 1B – see ESI† for further SEM images). The gel was also analysed by stress sweep rheometry. Gels were formed in a lipless glass vial and then transferred to the rheometer in flat plate geometry and allowed to stabilise. The samples reached a maximum elastic modulus G′ of 73![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Pa with G′′ of 2000 Pa. Gels yielded at a stress of 250 Pa. The fact that G′ is more than an order of magnitude greater than G′′ confirms the solid-like nature of the materials and confirms they are true gels (Fig. 1C).38
000 Pa with G′′ of 2000 Pa. Gels yielded at a stress of 250 Pa. The fact that G′ is more than an order of magnitude greater than G′′ confirms the solid-like nature of the materials and confirms they are true gels (Fig. 1C).38
The carboxylic acid residues on the OD/c-NC gels have the potential to interact strongly with the hydrogen bonding functionality of drug-like molecules, effectively locally immobilising the drug substances on the surface of the gel fibre and providing a nucleation surface. Accordingly the crystallization of a variety of examples of drugs and drug-like molecules bearing carboxylic acid, amide and nitrogen heterocyclic functionality within the novel DMSO OD/c-NC gel was examined. The targets chosen were dopamine hydrochloride, mexiletine hydrochloride, p-aminohippuric acid, benzocaine, sulfapyridine, L-valine, hydrochlorothiazole, 4-aminopyridine, 5-aminosalicylic acid, carisoprodol, isoniazid, ethionamide, sulfamethazine, sulfamerazine, sulfamethoxazole and sulfadiazine. As a control experiment identical crystallizations were also undertaken on pure DMSO without the gel. Mixtures of drug substance (10–500 mg) with 3 mg c-NC in the presence of OD were warmed in 1 mL DMSO and then allowed to cool to room temperature upon which gelation occurred. Gel formation was observed for all drug substances with the exception of 5-aminosalicylic acid and sulfathiazole which completely prevented gelation. In general many drugs weakened the gels. In the case of sulfapyridine and sulfamethoxazole this was confirmed by stress sweep rheometry with G′ values for the drug-gel mixture below 1000 Pa and yield stress of only 5 Pa in the presence of excess drug substance (see ESI,† Fig. S3). In the presence of very large excesses (e.g. 500 mg mL−1 of dopamine hydrochloride, mexiletine hydrochloride and aminohippuric acid) many drug substances completely inhibited gelation. We attribute this effect to factors such as increasing concentration of ions in solution and changes in pH disrupting the interactions between the gelators themselves and is analogous to the effect of added anions on the strength of supramolecular gels.39,40
In the case of sulfapyridine large, colourless block-like crystals formed within the gels upon standing at room temperature at concentrations in excess of 100 mg mL−1, however no crystallization occurred under the same conditions in the pure DMSO control experiments. These crystals were analysed by single crystal X-ray diffraction and proved to be a new 1:1 DMSO solvate (see ESI,† Fig. S4). Sulfapyridine is an extensively studied antibacterial discovered by May and Baker in 1937 and used in the second world war, not least to treat Winston Churchill's pneumonia.41 It exists in nine pure polymorphic forms42–44 and the CSD contains entries for 1:1 solvates with dioxane, tetrahydrofuran and nitromethane as well as a co-crystal with oxalic acid dibutyl ester and a piperidinium salt. The new form is isomorphous with the previously reported nitromethane solvate45 with the DMSO molecules situated in a discrete pocket in the structure arising from the V-shape of the sulfapyridine molecules, Fig. 2. The sulfapyridine is in the common imide form44 and forms hydrogen bonded dimers linked to the adjacent molecule by hydrogen bonding from the amino group to the sulfonyl oxygen atoms.
 | ||
| Fig. 2 X-ray crystal; structure of the 1:1 DMSO sulfapyridine solvate isolated from a nanocellulose gels showing the solvent pocket and principal hydrogen bonding interactions. | ||
While the structure itself is unremarkable, the fact that the c-NC gel reproducibly induces crystallization of such a well-studied compound from a solvent in which it does not crystallize in the absence of the gel indicates that the gel has a marked influence on the crystallization behaviour of the material. The equilibrium solubility of sulfapyridine in DMSO at room temperature is around 5 mM or 1.25 mg mL−1,46 and hence both gel and solution crystallizations are highly supersaturated. As a result the gel must be acting as a kinetic nucleation promoter rather than thermodynamically ‘salting out’ the solute.47
Crystals suitable for single crystal X-ray diffraction were also isolated from c-NC gels for the anti-tuberculosis drug isoniazid. The diffraction results revealed that this crystals are of the one known polymorph of isoniazid.48 The crystals were obtained from both the gel and the solution control experiments, however those from the gel were larger and better formed. The solution grown crystals adopt a needle-like habit, while the gel grown samples adopt a block morphology, Fig. 3, suggesting specific interactions of the growing crystal with the c-NC.
 | ||
| Fig. 3 (a) Needle crystals of isoniazid grown from DMSO solution and (b) block shaped crystals from DMSO OD/c-NC gels under the same conditions. | ||
Crystallization of another sulfa-antibiotic sulfamethoxazole from the OD/c-NC DMSO gels (see ESI,† Fig. S5) resulted in the formation of a surprising octadecylammonium salt of deprotonated sulfamethoxazole as a 1:2 DMSO solvate. The structure adopts an interdigitated lipid bilayer type arrangement with a polar region comprising DMSO solvent bridged sulfamethoxazole dimers, linked by hydrogen bonding from the amine NH groups to the DMSO oxygen atoms. The octadecylammonium ions interact with the sulfamethoxazole anions via hydrogen bonding to the sulfonyl oxygen atoms, Fig. 4. The crystallization of this large structure of a flexible long chain ammonium salt is relatively surprising. While crystals were not obtained in the absence of the c-NC gel it is not clear if the gel plays a direct role in the formation of this material. However, the structure does suggests a possible role for the ammonium-ion-sheathed gel in immobilising solutes by ion pairing via the formation of intercalated lipid bilayers on the fibre surface.
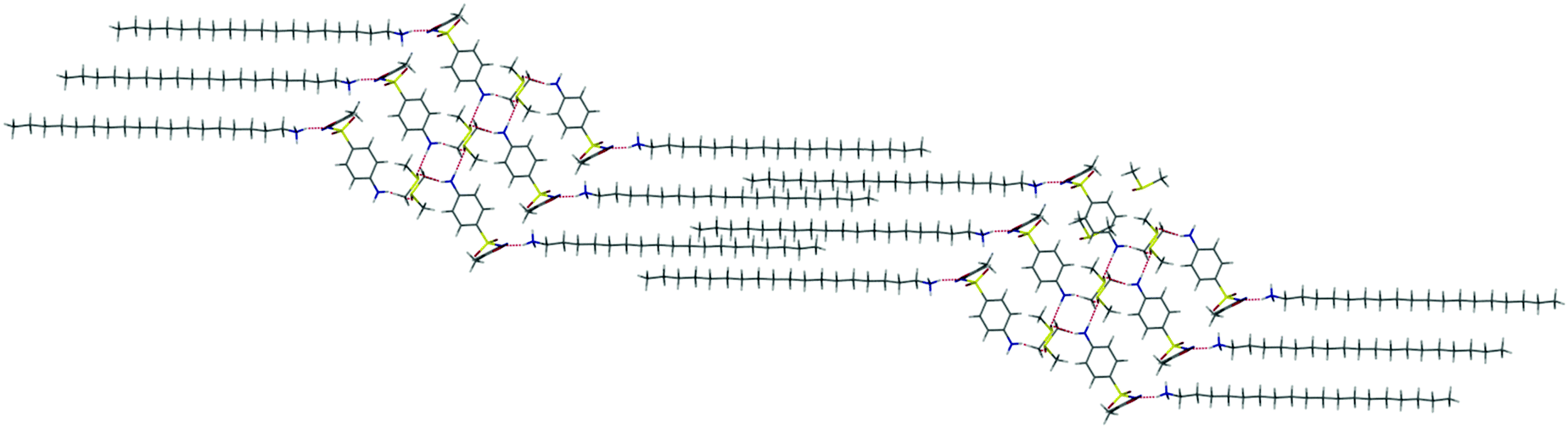 | ||
| Fig. 4 X-ray crystal structure of the octadecylammonium salt of deprotonated sulfamethoxazole 1:2 DMSO solvate isolated from a nanocellulose gels showing the solvent bilayer arrangement of the octadecylammonium ions. | ||
In a further attempt to develop novel NC based crystallization media we also prepared sulfonated nanocellulose (s-NC) according to the published procedure.49 The s-NC forms organogels in DMSO at 0.8 wt% (see ESI,† Fig. S6). These gels were used to crystallize sulfapyridine and also resulted in the formation of the new 1:1 DMSO solvate.
In conclusion we have shown that a readily available, hydrophilic nanomaterial can be modified to give an effective organogelator with low critical gelation concentration by addition of a cationic surfactant. The resulting hybrid organogels are suitable for use as a novel crystallization method for a range of pharmaceuticals. The effects of the gel are generally to eliminate convection effects and promote crystallization possibly by enhancing nucleation rate. Gel grown crystals are generally better formed. In the well-known systems studied here the gel medium has resulted in novel results, namely formation of a new solvate crystal form, habit modification and crystallization of a salt of a flexible long-chain surfactant. Novel organogel media offer considerable promise for application as part of an advanced solid forms pharmaceutical screening approach, particularly in the search for hard to nucleate or kinetically unfavourable solid forms.
We thank the Engineering and Physical Sciences Research Council and the Spanish Ministry of Innovation and Science for funding projects EP/J013021/1 and CTQ2014-52939R, respectively. This work was also supported by the IDEP (Institute of postgraduate studies). We also thank the Central Service for Research Support, SCAI (UCO), for its assistance in the NMR measurements.
Notes and references
- S. Byrn, R. Pfeiffer, M. Gany, C. Hoiberg and G. Poochikian, Pharm. Res., 1995, 12, 945 CrossRef CAS.
- A. J. Cruz-Cabeza and J. Bernstein, Chem. Rev., 2014, 114, 2170 CrossRef CAS PubMed.
- J. Bernstein, Cryst. Growth Des., 2011, 11, 632 CAS.
- J. Bernstein, Chem. Commun., 2005, 5007 RSC.
- K. M. Steed and J. W. Steed, Chem. Rev., 2015, 115, 2895 CrossRef CAS PubMed.
- G. P. Stahly, Cryst. Growth Des., 2007, 7, 1007 CAS.
- J. W. Steed, Trends Pharmacol. Sci., 2013, 34, 185 CrossRef CAS PubMed.
- N. Blagden, S. J. Coles and D. J. Berry, CrystEngComm, 2014, 16, 5753 RSC.
- N. Blagden and R. J. Davey, Cryst. Growth Des., 2003, 3, 873 CAS.
- E. H. Lee, Asian J. Pharm. Sci., 2014, 9, 163 CrossRef.
- Polymorphism in Pharmaceutical Solids, ed. H. G. Brittain, Marcel Dekker Inc., New York, 1999 Search PubMed.
- W. Cabri, P. Ghetti, G. Pozzi and M. Alpegiani, Org. Process Res. Dev., 2007, 11, 64 CrossRef CAS.
- J. Bauer, S. Spanton, R. Henry, J. Quick, W. Dziki, W. Porter and J. Morris, Pharm. Res., 2001, 18, 859 CrossRef CAS.
- J. K. Guillory, in Polymorphism in Pharmaceutical Solids, ed. H. G. Brittain, Marcel Dekker Inc., New York, 1999, pp. 183–226 Search PubMed.
- M. A. Neumann, J. van de Streek, F. P. A. Fabbiani, P. Hidber and O. Grassmann, Nat. Commun., 2015, 6, 7793 CrossRef CAS PubMed.
- S. L. Price, Chem. Soc. Rev., 2014, 43, 2098 RSC.
- S. L. Morissette, Ö. Almarsson, M. L. Peterson, J. F. Remenar, M. J. Read, A. V. Lemmo, S. Ellis, M. J. Cima and C. R. Gardner, Adv. Drug Delivery Rev., 2004, 56, 275 CrossRef CAS PubMed.
- C. E. Nicholson, C. Chen, B. Mendis and S. J. Cooper, Cryst. Growth Des., 2011, 11, 363 CAS.
- C. Chen, O. Cook, C. E. Nicholson and S. J. Cooper, Cryst. Growth Des., 2012, 11, 2228 Search PubMed.
- Y. Diao, K. E. Whaley, M. E. Helgeson, M. A. Woldeyes, P. S. Doyle, A. S. Myerson, T. A. Hatton and B. L. Trout, J. Am. Chem. Soc., 2012, 134, 673 CrossRef CAS PubMed.
- Q. Jiang and M. D. Ward, Chem. Soc. Rev., 2014, 43, 2066 RSC.
- J.-B. Arlin, L. S. Price, S. L. Price and A. J. Florence, Chem. Commun., 2011, 47, 7074 RSC.
- J. A. Foster, M.-O. M. Piepenbrock, G. O. Lloyd, N. Clarke, J. A. K. Howard and J. W. Steed, Nat. Chem., 2010, 2, 1037 CrossRef CAS PubMed.
- A. Dawn, K. S. Andrew, D. S. Yufit, Y. X. Hong, J. P. Reddy, C. D. Jones, J. A. Aguilar and J. W. Steed, Cryst. Growth Des., 2015, 15, 4591 CAS.
- F. Aparicio, E. Matesanz and L. Sánchez, Chem. Commun., 2012, 48, 5757 RSC.
- L. A. Estroff, L. Addadi, S. Weiner and A. D. Hamilton, Org. Biomol. Chem., 2004, 2, 137 CAS.
- G. O. Lloyd, M. O. M. Piepenbrock, J. A. Foster, N. Clarke and J. W. Steed, Soft Matter, 2012, 8, 204 RSC.
- M.-O. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. W. Steed, Chem. Rev., 2010, 110, 1960 CrossRef CAS PubMed.
- K. W. K. Tong, S. Dehn, J. E. A. Webb, K. Nakamura, F. Braet and P. Thordarson, Langmuir, 2009, 25, 8586 CrossRef CAS PubMed.
- D. K. Kumar and J. W. Steed, Chem. Soc. Rev., 2014, 43, 2080 RSC.
- C. Ruiz-Palomero, M. L. Soriano and M. Valcárcel, J. Chromatogr. A, 2016, 1428, 352 CrossRef CAS PubMed.
- C. Ruiz-Palomero, M. L. Soriano and M. Valcárcel, Talanta, 2014, 125, 72 CrossRef CAS PubMed.
- D. Klemm, F. Kramer, S. Moritz, T. Lindstrom, M. Ankerfors, D. Gray and A. Dorris, Angew. Chem., Int. Ed., 2011, 50, 5438 CrossRef CAS PubMed.
- I. Siro and D. Plackett, Cellulose, 2010, 17, 459 CrossRef CAS.
- C. Ruiz-Palomero, M. L. Soriano and M. Valcárcel, Sens. Actuators, B, 2016, 229, 31 CrossRef CAS.
- T. Saito, Y. Nishiyama, J.-L. Putaux, M. Vignon and A. Isogai, Biomacromolecules, 2006, 7, 1687 CrossRef CAS PubMed.
- G. Cravotto and P. Cintas, Chem. Soc. Rev., 2009, 38, 2684 RSC.
- G. Yu, X. Yan, C. Han and F. Huang, Chem. Soc. Rev., 2013, 42, 6697 RSC.
- M. O. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. W. Steed, Chem. Commun., 2008, 2644 RSC.
- G. O. Lloyd and J. W. Steed, Nat. Chem., 2009, 1, 437 CrossRef CAS PubMed.
- J. Lesch, The First Miracle Drugs, Oxford University Press, Oxford, 2007 Search PubMed.
- I. Bar and J. Bernstein, J. Pharm. Sci., 1985, 74, 255 CrossRef CAS PubMed.
- J. Bernstein, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1988, 44, 900 CrossRef.
- T. Gelbrich, T. L. Threlfall, A. L. Bingham and M. B. Hursthouse, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2007, 63, o323 CAS.
- M. M. Ghorab, M. S. Al-Said, H. A. Ghabbour, S. Chantrapromma and H.-K. Fun, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, o1030 CAS.
- Sulfapyridine materials data sheet, accessed 6/4/16, http://www.abcam.com/Sulfapyridine-ab145656.pdf.
- S. A. Miller, D. D. Dykes and H. F. Polesky, Nucleic Acids Res., 1988, 16, 1215 CrossRef CAS PubMed.
- A. Lemmerer, CrystEngComm, 2012, 14, 2465 RSC.
- J. H. Lee, S. H. Park and S. H. Kim, Macromol. Res., 2013, 21, 1218 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental data, images of gels and crystals, DSC and rheology plots as well as details of single crystal structure determination in CIF format. The underlying research data for this paper is available in accordance with EPSRC open data policy from DOI: http://dx.doi.org/10.15128/m900nt40f. CCDC 1473482 and 1473483. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc03088b |
| This journal is © The Royal Society of Chemistry 2016 |