
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuinonediimines as redox-active organocatalysts for oxidative coupling of aryl- and alkenylmagnesium compounds under molecular oxygen†
Toru
Amaya
*,
Riyo
Suzuki
and
Toshikazu
Hirao‡
*
Department of Applied Chemistry, Graduate School of Engineering, Osaka University, Yamada-oka, Suita, Osaka 565-0871, Japan. E-mail: amaya@chem.eng.osaka-u.ac.jp; hirao@chem.eng.osaka-u.ac.jp
First published on 12th May 2016
Abstract
It is revealed that N,N′-diphenyl-p-benzoquinonediimine works as a redox-active organocatalyst for the oxidative homo-coupling of aryl- and alkenylmagnesium compounds under molecular oxygen. The catalytic cycle was formally monitored by 1H NMR experiments.
Organocatalysis is now one of the most thriving areas in organic synthesis.1 The reaction focused on here is organocatalytic oxidative homo-coupling of aryl- and alkenylmagnesium compounds under molecular oxygen (Scheme 1). However, such a catalyst has been rarely developed before (only a few examples have been reported as described later) despite the upsurge of interest in organocatalysis.
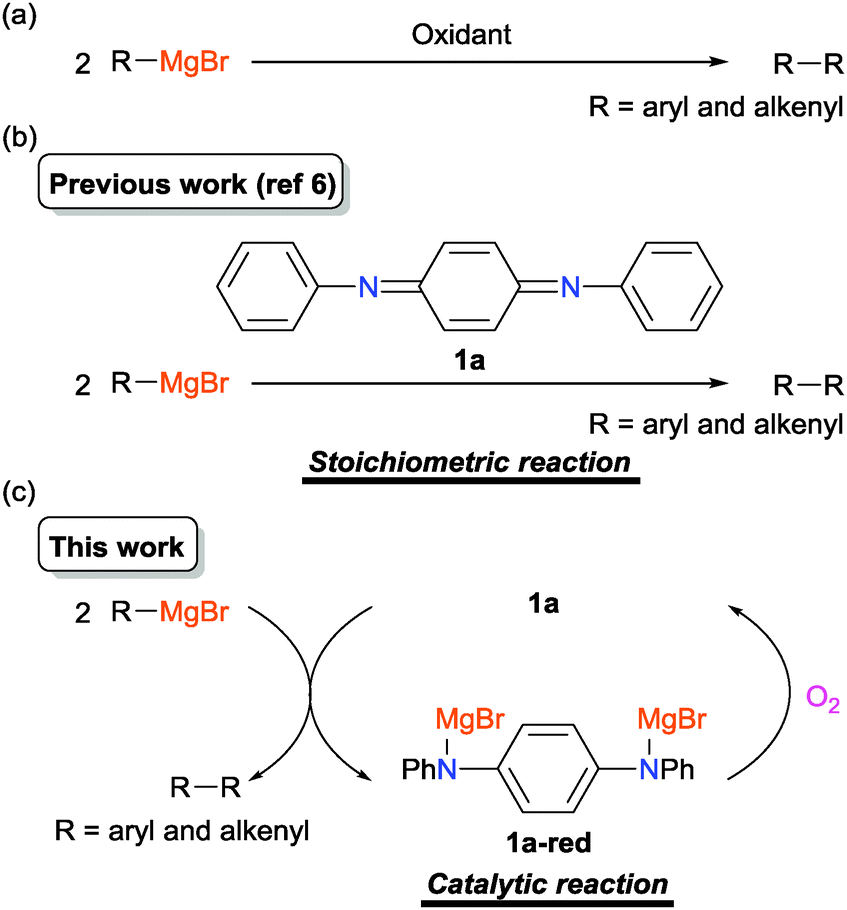 | ||
| Scheme 1 (a) Oxidative homo-coupling of aryl- and alkenylmagnesium compounds. (b) Our previous study on the stoichiometric reaction. (c) This work: organocatalytic oxidative homo-coupling using quinonediimine 1a as a redox-active catalyst under molecular oxygen. | ||
The oxidative homo-coupling of aryl- and alkenylmagnesium compounds (Scheme 1a) is one of the efficient methods for the synthesis of symmetrical biaryls and bialkenyls. Stoichiometric use of high-valent transition metal oxidants generally works well for this reaction, which has been studied since about a century ago.2 In the last decade, some catalytic systems using transition metals have been constructed.3 Examples for the combination of the catalyst and the terminal oxidant are shown below: FeCl3 or MnCl2-1,2-dihaloethane,3a–c Li2CuCl4–CuBr·SMe2-di-tert-butyldiaziridinone,3d FeCl3 or MnCl2 or Li2CuCl4 or RuCl(C3S5)H2O(PPh3)2–O2,3e–h and FeCl3 or MnCl2–N2O.3i On the other hand, the methodology employing organic oxidants is far more behind in this area, and the research studies have been quite limited.4 One of the reasons is the difficulty in making the electron-transfer type reaction take place in preference to the nucleophilic addition in a competitive situation, for example employing stoichiometric 1,4-benzoquinone as an oxidant in this case.4d In 2006, Mayr, Knochel and co-workers proved the utility of 3,3′,5,5′-tetra-tert-butyldiphenoquinone with bulky tert-butyl groups in both sides of carbonyls as a stoichiometric oxidant for the coupling reaction.4c This finding should be a milestone in this area from the viewpoint of metal-free coupling. The next challenge should be the construction of the organocatalytic system under molecular oxygen, which is one of the desirable terminal oxidants in the catalytic oxidation reactions from the viewpoint of sustainable chemistry. However, a possible side reaction, that is reaction of the arylmagnesium compounds with molecular oxygen, makes this reaction more complicated. Only two examples have been reported for the organocatalytic oxidative coupling of aryl- and alkenylmagnesium compounds. One is a 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO) radical catalyzed reaction reported by Studer and co-workers.4e The other is a recently reported 3,4,5-trifluoro-1,2-bis(perfluorophenyl)cyclopentadienyl anion-catalyzed reaction by Korenaga and co-workers.4f In our previous study, oxidation catalysts consisting of redox-active poly- and oligoaniline derivatives were developed,5 including catalysts for oxidative coupling of phenol derivatives using molecular oxygen as a terminal oxidant.5c Recently, it was also revealed that N,N′-diphenyl-p-benzoquinonediimine (1a) induces the stoichiometric oxidative coupling of aryl- and alkenylmagnesium compounds (Scheme 1b).6 Here, we report the catalytic version of the reaction under molecular oxygen as a terminal oxidant (Scheme 1c).
The investigation began with the oxidative homo-coupling of (1-phenylvinyl)magnesium bromide (2a) in the presence of a catalytic amount of quinonediimine 1a under molecular oxygen. The employed 1a was prepared by the oxidation of N,N′-diphenyl-p-phenylenediamine with iodosylbenzene under metal free conditions. The ICP-AES experiment of 1a certified that the contamination of transition metals such as Cu, Fe, Mn and Pd is negligibly small for this reaction (Cu, Fe and Mn: limit of quantification, Pd: 6 × 10−5 mol% to 2). A THF solution of 2a was added to quinonediimine 1a in THF under molecular nitrogen at room temperature. After 5 min, molecular oxygen was flowed through the flask for 30 min. The desired coupling product 3a was formed in 64% yield, where the turnover number of 1a is 3.2 because 1a is a two-electron oxidant (Scheme 2). Acetophenone was also formed in 8% as a side product. As a control experiment, the same reaction was carried out in the absence of 1a. The yield of 3a was 6%.
 | ||
| Scheme 2 1a-catalyzed oxidative coupling of 2a and its control experiment. | ||
The reaction was monitored with time by sampling (Fig. 1). First, the reaction took place under molecular nitrogen, where the red colour of 1a changed to pale yellow. Flowing molecular oxygen (5–12 min) made the colour of the reaction mixture change to green and then red. In this period, an increase in the yield was observed. The red colour suggests the regeneration of 1a. After the colour of the reaction mixture turned red, an increase of the yield was almost stopped. This suggests that (1-phenylvinyl)magnesium bromide (2a) was almost consumed.
 | ||
| Fig. 1 Profile of the yield for 3a with time. | ||
Increasing the reaction temperature to 60 °C gave the product 3a in 72% yield (Scheme 2). Bubbling of molecular oxygen instead of flowing did not induce an increase in the yield. The reaction under dry air instead of molecular oxygen gave 3a in 68% yield. Decreasing the amount of 1a to 5 mol% lowered the yield to 47%, whereas the turnover number of 1a increased to 4.7.
Some quinonediimine derivatives 1b–d7 were also tested as catalysts instead of 1a for this reaction. However, better results were not obtained as compared to that with 1a (Table 1, entries 1–3). In the case of 1b, the reaction proceeded catalytically although the efficiency was low (Table 1, entry 1). On the other hand, there seems to be a problem in the regeneration step in the case of 1c (Table 1, entry 2). Catalyst 1d did not give rise to the oxidative coupling of 2a even in the stoichiometric reaction (Table 1, entry 3).

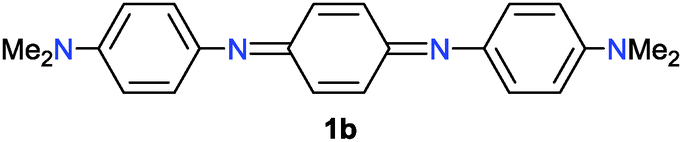

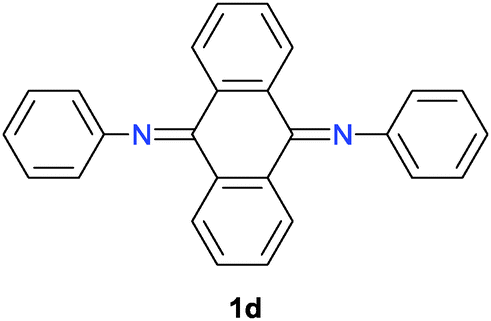
Table 2 shows the screening of the solvents for this reaction. The reaction was performed at room temperature. Use of Et2O, CH2Cl2 and cyclopentyl methyl ether instead of THF was not effective for this reaction (Table 2, entries 2–4). The mixed solution of THF/CH2Cl2 and THF/cyclopentyl methyl ether also did not give the better results than THF (Table 2, entries 5 and 6).
|
|
||
|---|---|---|
| Entry | Solvent | Yielda/% |
| a Yield was calculated by the integral ratio of the peaks for 3a and 1,3,5-trimethoxybenzene as an internal standard in the 1H NMR spectrum of the crude mixture. b This entry is the same as a result in Scheme 2. | ||
| 1b | THF | 64 |
| 2 | Et2O | 42 |
| 3 | CH2Cl2 | 20 |
| 4 | Cyclopentyl methyl ether | 36 |
| 5 | CH2Cl2/THF | 64 |
| 6 | Cyclopentyl methyl ether/THF | 54 |

To gain information on the reaction path, the reaction was followed by 1H NMR spectroscopy (Fig. 2, and the enlarged figure is shown in Fig. S1, ESI†). At first, 1a is dissolved in THF-d8, the spectrum is shown in Fig. 2a. To the solution was added a stoichiometric amount of 2a (200 mol%) under molecular nitrogen. The peaks for 1a disappeared and the peaks for the homo-coupling product 3a appeared with the reduced quinonediimine compound 1a-red (Fig. 2b). The broad signals of 1a-red well-accorded with those for the separately prepared one by the reaction of N,N′-diphenyl-p-phenylenediamine with phenylmagnesium bromide (see the supporting information of ref. 6). The broadening seems to be due to aggregation. Introduction of molecular oxygen clearly showed the regeneration of 1a (Fig. 2c). To the mixture was added a stoichiometric amount of 2a (200 mol%), again resulting in the complete consumption of 1a and the formation of 3a and 1a-red (Fig. 2d). These experiments formally show the catalytic cycle based on 1a and 1a-red with molecular oxygen as a terminal oxidant.
 | ||
| Fig. 2 1H NMR experiment to follow the redox cycle of 1a in the oxidative coupling of 2a and reoxidation under molecular oxygen. | ||
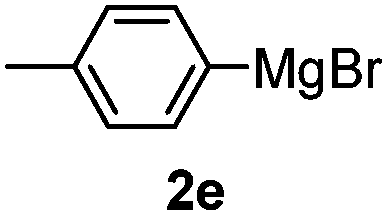
Table 3 shows the substrate scope for the 1a-catalyzed oxidative homo-coupling reaction.§ For the reaction of arylmagnesium compounds 2b–h, use of dry air instead of molecular oxygen in the presence of 15 mol% of 1a gave the better results (Table 3, entries 1–7). Substitution of the benzene ring with p-MeO-, p-F-, p-Me- and o-Me was not a problem and gave the corresponding homo-coupling products 3c–f in good yields (70–79%, Table 3, entries 2–5). Bulky mesitylmagnesium bromide (2g) also homo-coupled to give product 3g in 71% yield (Table 3, entry 6). The coupling of 2-naphthylmagnesium bromide (2h) showed the best yield (85%, Table 3, entry 7). The reaction of (1-arylvinyl)magnesium bromides 2i–j took place under molecular oxygen to provide the products 3i–j in moderate yields (Table 3, entries 8 and 9).
|
|
|||
|---|---|---|---|
| Entry | Substrate | Product | Yielda/% |
| a Isolated yield. b Including a small amount of the impurity. c 10 mol% of 1a was used and the reaction was conducted under molecular oxygen. d Yield was calculated by the integral ratio of the peaks for 3a and 1,3,5-trimethoxybenzene as an internal standard in the 1H NMR spectrum of the crude mixture. | |||
| 1 |

|

|
71 |
| 2 |
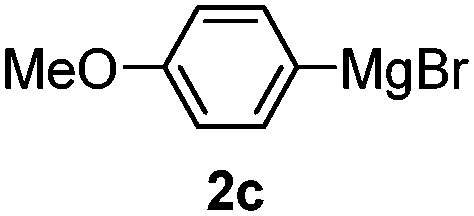
|

|
79 |
| 3 |
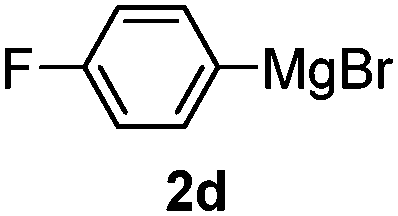
|

|
74b |
| 4 |
|

|
77 |
| 5 |

|

|
70 |
| 6 |

|

|
71 |
| 7 |

|

|
85 |
| 8c |

|

|
70d |
| 9c |
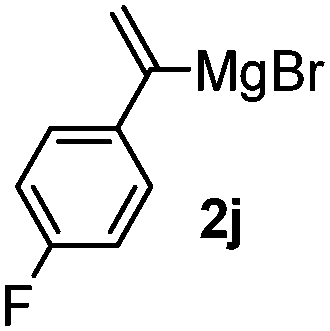
|
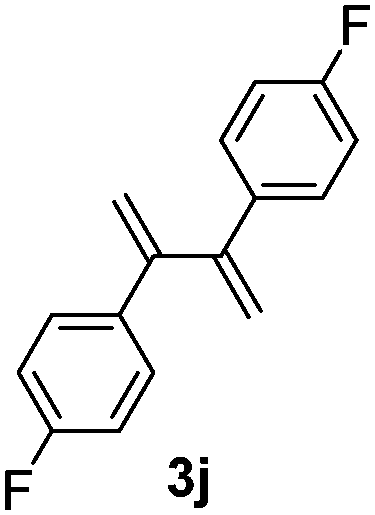
|
61d |

In conclusion, it is revealed that quinonediimine 1a works as a redox-active organocatalyst for the oxidative homo-coupling of aryl- and alkenylmagnesium compounds under molecular oxygen. The catalytic cycle was formally monitored by 1H NMR experiments. It should be noted that this organocatalyst can catalyze the oxidative carbon–carbon bond formation using molecular oxygen as a terminal oxidant, which is one of the challenging topics in the field of organocatalysts.
This work was supported by a Grant-in-Aid for Scientific Research on Innovative Areas “Advanced Molecular Transformations by Organocatalysts” from The Ministry of Education, Culture, Sports, Science and Technology, Japan (26105736).
Notes and references
- D. W. C. MacMillan, Nature, 2008, 455, 304 CrossRef CAS PubMed.
- (a) G. M. Bennett and E. E. Turner, J. Chem. Soc., Trans., 1914, 105, 1057 RSC; (b) J. Krizewsky and E. E. Turner, J. Chem. Soc., Trans., 1919, 115, 559 RSC; (c) J. H. Gardner and P. Borgstrom, J. Am. Chem. Soc., 1929, 51, 3375 CrossRef CAS; (d) C. C. Vernon, J. Am. Chem. Soc., 1931, 53, 3831 CrossRef CAS; (e) H. Gilman and M. Lichtenwalter, J. Am. Chem. Soc., 1939, 61, 957 CrossRef CAS; (f) M. S. Kharasch and E. K. Fields, J. Am. Chem. Soc., 1941, 63, 2316 CrossRef CAS; (g) A. Mckillop, L. F. Elsom and E. C. Taylor, Tetrahedron, 1970, 26, 4041 CrossRef CAS; (h) A. Inoue, K. Kitagawa, H. Shinokubo and K. Oshima, Tetrahedron, 2000, 56, 9601 CrossRef CAS.
- (a) T. Nagano and T. Hayashi, Org. Lett., 2005, 7, 491 CrossRef CAS PubMed; (b) G. Cahiez, C. Chaboche, F. Mahuteau-Betzer and M. Ahr, Org. Lett., 2005, 7, 1943 CrossRef CAS PubMed; (c) Z. Zhou and W. Xue, J. Organomet. Chem., 2009, 694, 599 CrossRef CAS; (d) Y. Zhu, T. Xiong, W. Han and Y. Shi, Org. Lett., 2014, 16, 6144 CrossRef CAS PubMed; (e) G. Cahiez, A. Moyeux, J. Buendia and C. Duplais, J. Am. Chem. Soc., 2007, 129, 13788 CrossRef CAS PubMed; (f) W. Liu and A. Lei, Tetrahedron Lett., 2008, 49, 610 CrossRef CAS; (g) G. Cahiez, C. Duplais and J. Buendia, Angew. Chem., Int. Ed., 2009, 48, 6731 CrossRef CAS PubMed; (h) S.-K. Hua, Q.-P. Hu, J. Ren and B.-B. Zeng, Synthesis, 2013, 45, 518 CrossRef CAS; (i) G. Kiefer, L. Jeanbourquin and K. Severin, Angew. Chem., Int. Ed., 2013, 52, 6302 CrossRef CAS PubMed.
- (a) J.-W. Cheng and F.-T. Luo, Tetrahedron Lett., 1988, 29, 1293 CrossRef CAS; (b) T. Nishiyama, T. Seshita, H. Shodai, K. Aoki, H. Kameyama and K. Komura, Chem. Lett., 1996, 549 CrossRef CAS; (c) A. Krasovskiy, A. Tishkov, V. del Amo, H. Mayr and P. Knochel, Angew. Chem., Int. Ed., 2006, 45, 5010 CrossRef CAS PubMed; (d) T. Ramnial, S. A. Taylor, J. A. C. Clyburne and C. J. Walsby, Chem. Commun., 2007, 2066 RSC; (e) M. S. Maji, T. Pfeifer and A. Studer, Angew. Chem., Int. Ed., 2008, 47, 9547 CrossRef CAS PubMed; (f) T. Korenaga, K. Nitatori, H. Muraoka, S. Ogawa and K. Shimada, Org. Lett., 2015, 17, 5500 CrossRef CAS PubMed.
- (a) T. Hirao, M. Higuchi, I. Ikeda and Y. Ohshiro, J. Chem. Soc., Chem. Commun., 1993, 2, 194 RSC; (b) M. Higuchi, I. Ikeda and T. Hirao, J. Org. Chem., 1997, 62, 1072 CrossRef CAS; (c) T. Hirao and S. Fukuhara, J. Org. Chem., 1998, 63, 7534 CrossRef CAS PubMed ; for accounts: ; (d) T. Amaya and T. Hirao, Synlett, 2011, 435 CAS; (e) T. Moriuchi and T. Hirao, Acc. Chem. Res., 2012, 45, 347 CrossRef CAS PubMed.
- T. Amaya, R. Suzuki and T. Hirao, Chem. – Eur. J., 2014, 20, 653 CrossRef CAS PubMed.
- For the preparation of (a) 1b: Y. Wei, C. Yang and T. Ding, Tetrahedron Lett., 1996, 37, 731 CrossRef CAS; (b) 1c: H. W. Boone, J. Bryce, T. Lindgren, A. B. Padias and H. K. Hall, Jr., Macromolecules, 1997, 30, 2797 CrossRef CAS; (c) 1d: H. K. Hall, Jr., A. B. Padias, P. A. Williams, J.-M. Gosau, H. W. Boone and D.-K. Park, Macromolecules, 1995, 28, 1 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc03053j |
| ‡ Present address: The Institute of Scientific and Industrial Research, Osaka University, Mihoga-oka, Ibaraki, Osaka 567-0047, Japan. |
| § A representative procedure: To a two-neck 10 mL dried flask with a condenser was added 1a (39.4 mg, 0.15 mmol), and the atmosphere was replaced by molecular nitrogen. Dry THF (1.0 mL) was added. A 0.50 M THF solution of 2-naphthylmagnesium bromide (2h) (2.0 mL, 1.0 mmol) was dropwise added at room temperature. After stirring for 5 min at room temperature, the flask was put in a pre-warmed oil bath (60 °C). Then, air dried through concentrated H2SO4 was flowed slowly (8.3 mL min−1) for 55 min. The reaction mixture was stirred at 60 °C in this period, and then quenched with a mixed aqueous solution of NaHCO3/Na2S2O3. To the mixture was added CH2Cl2. The aqueous layer was filtered through filter paper and extracted with CH2Cl2 three times. The combined organic layer was washed with brine and dried with Na2SO4. After filtration, the mixture was concentrated in vacuo. The residue was purified by silica-gel chromatography (0 to 10% CH2Cl2 in hexane) to give 3h as a white flaky solid (108.5 mg, 0.43 mmol, 85%). |
| This journal is © The Royal Society of Chemistry 2016 |