
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Preparation of difluoromethylthioethers through difluoromethylation of disulfides using TMS-CF2H†
Joseph L.
Howard
,
Christiane
Schotten
,
Stephen T.
Alston
and
Duncan L.
Browne
*
School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: dlbrowne@cardiff.ac.uk
First published on 7th June 2016
Abstract
We report an operationally simple, metal-free approach for the late-stage introduction of the important lipophilic hydrogen-bond donor motif, SCF2H. This reaction converts diaryl- and dialkyl-disulfides into the corresponding aryl/alkyl–SCF2H through the nucleophilic transfer of a difluoromethyl group with good functional group tolerance. This method is notable for its use of commercially available TMSCF2H, and does not rely on the need for handling of sensitive metal complexes.
The fluorination of organic molecules continues to lead to materials with improved properties with which to fuel our modern society. As testament to the dramatic improvements available by the fluorination of organic materials, recent years have seen a growth, at the discovery phase, of a late-stage fluorine scanning approach.1 Such an approach looks to fine-tune physicochemical properties by the inclusion of fluorine atoms.2 Whilst some effects of fluorination can be rationally predicted and thus provide a toolbox to guide fine-tuning, several key observations have not been predictable, but have lead to exciting new observations for organo-fluorine chemistry.3 It is perhaps owing to this rich-seam of uncharted beneficial effects that the synthesis community are increasingly interested in organo-fluorine methods. In contrast to this late-stage approach however, there is now increasing pressure to reduce the amount of fluorinated waste materials making it into waste streams and eventually in to the ecosystem. Especially given that degradation of fluorous compounds through standard biological processes is greatly retarded by nature's poor ability to process fluorinated materials.4 As a fine balance between this dichotomy we are interested in developing rationally designed methods for late stage fluorination approaches to under-represented fluorous motifs. Herein our particular focus has been on the series consisting of fluorinated methylthioethers R-SCFxH(3−x) (Fig. 1). Initially we considered the x = 0 state; methylthioethers. This motif is rarely present in biologically active materials, owing to a poor metabolic profile characterised by oxidation at sulfur by cytochromes (such as P450). The resultant sulfoxides/sulfones are then more easily cleared before the target is reached. Notably, stable sulfoxides and sulfones are not uncommonly found in biologically active materials.5 Assessment of the fully fluorinated variant; x = 3 state, highlights several examples of this motif present in successful-to-market biologically active materials.6 From an electronics perspective the sulfur atom in this case is more electron deficient (than in the x = 0 case) so the rate of oxidation through a nucleophilic at sulfur mechanism is greatly reduced (comparatively), resulting in reduced metabolism and reduced clearance levels. This leads to an ability for these molecules to reach the intended target before being metabolically cleared. This is however an over simplified view as the cLog
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P or liphophilicity of SCF3 is also different (greater) from that of SCH3.7 Increased lipophilicity manifests in to a number of phenomena, not the least of which include less specific binding to enzymes and better transport across the blood:brain barrier. Regarding the x = 2 state, the SCF2H motif maintains many of the properties displayed by SCF3, but, in addition, gains hydrogen-bond donor capabilities. Thus SCF2H essentially serves as a lipophilic hydrogen-bond donor motif for drug discovery, however the range of methods for the preparation of this motif are relatively narrow (compared to SCF3).
P or liphophilicity of SCF3 is also different (greater) from that of SCH3.7 Increased lipophilicity manifests in to a number of phenomena, not the least of which include less specific binding to enzymes and better transport across the blood:brain barrier. Regarding the x = 2 state, the SCF2H motif maintains many of the properties displayed by SCF3, but, in addition, gains hydrogen-bond donor capabilities. Thus SCF2H essentially serves as a lipophilic hydrogen-bond donor motif for drug discovery, however the range of methods for the preparation of this motif are relatively narrow (compared to SCF3).
 | ||
| Fig. 1 Effect of fluorine addition on thioethers. | ||
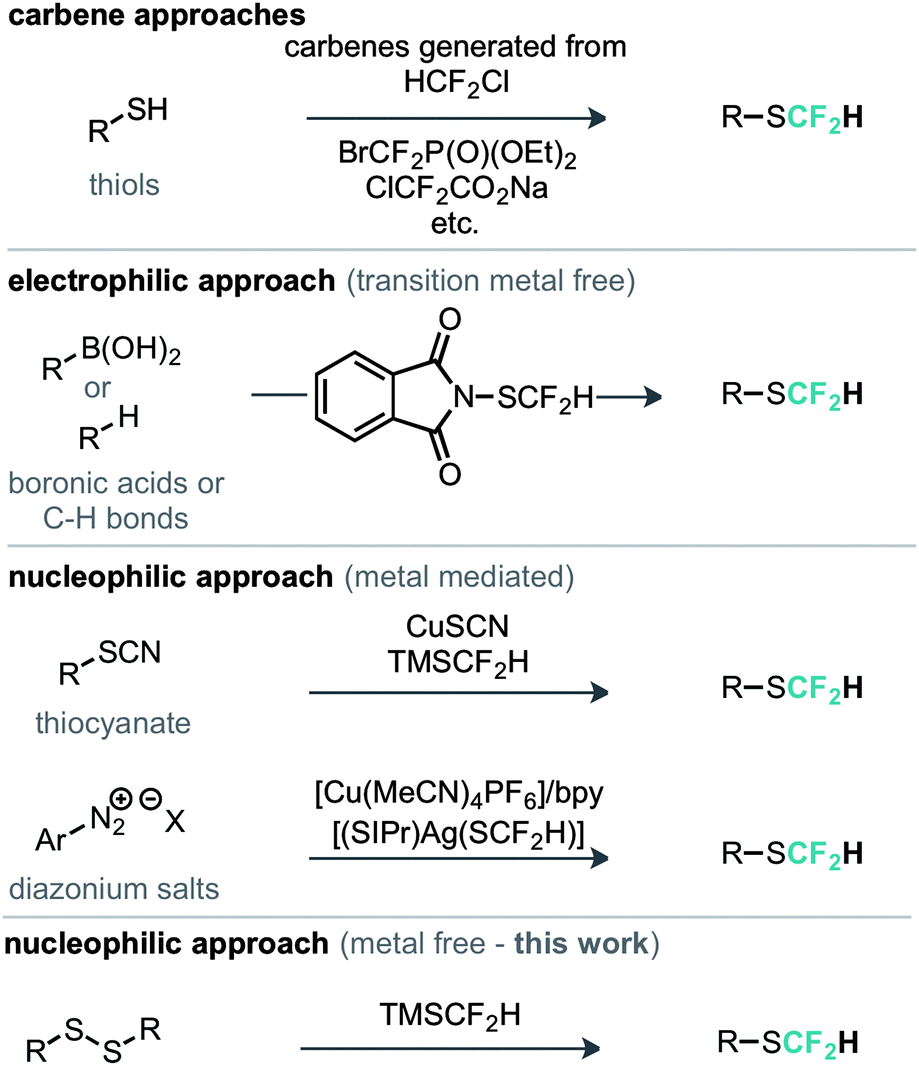
The most commonly explored approach to the SCF2H motif features the in situ generation of difluorocarbene followed by nucleophilic attack from a thiol or thiolate and protonation of the resulting difluoromethide (Scheme 1).8 With increasing attention in this moiety there have been recent examples of methods developed for its late stage introduction. For example an electrophilic strategy was reported, whereby N-difluoromethylthiophthalimide was demonstrated for the direct difluoromethylthiolation of a range of nucleophilic substrates such as boronic acids and aromatic C–H bonds.9 Shen has also developed a metal mediated nucleophilic protocol for the conversion of aromatic diazonium salts in to the corresponding aryl–SCF2H compounds using [(SIPr)Ag(SCF2H)] as the SCF2H source.10
 | ||
| Scheme 1 Synthetic routes to difluoromethylthioethers. | ||
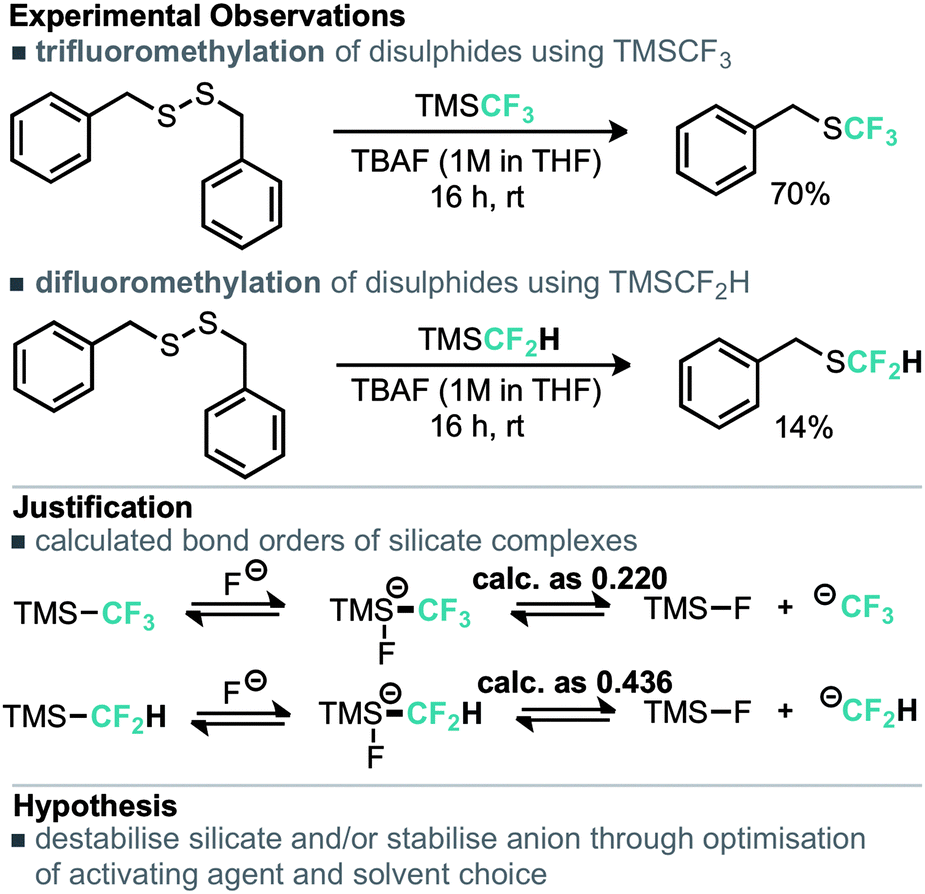
An alternative copper mediated approach was reported for the conversion of thiocyanates or aryl diazonium salts using TMSCF2H.11 Our approach was to develop an operationally simple method, whereby transfer of the required fluorinated carbon unit from a silicate complex to a disulfide electrophile would result in formation of the difluoromethylthioether. Indeed, during the course of our studies, very recently Zhang, Zhu and co-workers published on an identical strategy.12 Notably an analogous approach exists for trifluoromethylation of disulfides as reported by Langlois.13 In this instance the Ruppert–Prakash reagent, activated by fluoride from TBAF was shown to transfer to the corresponding benzylic disulfides in good yield. It was noted that aromatic disulfides offer poor conversion under these conditions with a single example reported (Scheme 2). For the difluoromethylthioether we initially commenced by simply repeating this approach with TMSCF2H. Under these conditions, a poor conversion of 14% was found and a control experiment in our hands highlighted that the work of Langlois was perfectly reproducible. This observation suggested that the lower conversion for –CF2H variants is inherent in the reactivity of these species. Indeed a report from Fuchikami describes the stability of both the TMSCF3 and the TMSCF2H fluoro–silicate complexes.14 Notably the authors calculate that the bond order (a reflection on the ion formation potential) for TMSCF3 was approximately half that of TMSCF2H, at 0.220, implying that the difluoromethylsilicate complex was less prone to generating the required difluoromethyl anion species. We hypothesised that appropriate choice of activating agent and solvent could help to destabilise the silicate complex and/or stabilise the desired ion formation, which would lead to improved nucleophile transfer. Our studies commenced by treating a solution of dibenzyldisulfide and TMSCF2H in THF with a range of activating agents (Table 1). Notably a range of fluoride sources, t-BuOK (previously shown to effectively activate TMSCF2H),15 and a combination of either of these with copper(I) salts failed to provide much conversion to the desired product. Upon switching to different solvents, it was found that simple fluoride sources could perform the required activation with CsF out-performing KF in both acetonitrile and N,N-dimethylacetamide (cf. entries 7, 8 and entries 9, 10, Table 1). Indeed, further solvent probing with CsF highlighted N-methylpyrrolidine as optimal, affording the desired product in 63% conversion. An increase in reaction temperature resulted in a poorer reaction yield, (Table 1, entry 14, 36%). Optimal conditions were reached by increasing the equivalents of TMSCF2H and CsF further (entry 18, 82% yield). With optimal conditions in hand for the metal-free nucleophilic difluoromethylation of dibenzyldisulfide we then turned attention to the generality of the scope of this method with respect to dialkyldisulfides. We evaluated several methods for the reliable and rapid synthesis of disulfide starting materials and found treatment of thiols with inexpensive dibromodimethylhydantoin to be the most effective.16 Pleasingly, a range of dibenzyldisulfides underwent the difluoromethylation reaction to provide the products in good yield (Scheme 3), including the ortho-bromo derivative 3 (73% yield). Non-benzylic substrates also participated in the reaction, including secondary alkyl disulfides, with the cyclopentyl variant 5 proceeding in 34% yield. Free alcohols are also amenable under the present methodology, with the difluoromethylthioether 6 undergoing reaction in 49% yield. However, in stark contrast to the trifluoromethylation approach reported by Langlois, the present method of difluoromethylation was highly effective on diaryldisulfides. As shown in Scheme 4, diaryldisulfides convert to the desired fluorinated products with good to excellent conversion. Both electron rich and electron poor examples proceed well, as do ortho-substituted systems and heteroaromatics, with a 2-pyridyl example converting to the SCF2H product (17) in 49% yield. Notably, whilst we have proven that isolation and accurate depiction of isolated yields is somewhat hampered by compound volatility, we believe that the late stage fluorination of more advanced drug-like scaffolds (higher molecular weight) would permit ready isolation of the SCF2H material as testament to this compounds 10 and 18 have been found to be isolable.17
 | ||
| Scheme 2 Project design and hypothesis. | ||
|
|
|||
|---|---|---|---|
| Entry | Activating agent (equiv.) | Solvent | Yielda (%) |
| a Yields calculated using 19F NMR spectroscopy with internal standard. b Reaction conducted at 60 °C. c 4 equiv. of TMSCF2H used. | |||
| 1 | TBAF (2.0) | THF | 14 |
| 2 | CsF (2.0) | THF | 1 |
| 3 | KF (2.0) | THF | 0 |
| 4 | t-BuOK (2.0) | THF | 5 |
| 5 | CuI/t-BuOK (2.0) | THF | 4 |
| 6 | CuI/CsF (2.0) | THF | 0 |
| 7 | KF (2.0) | MeCN | 0 |
| 8 | CsF (2.0) | MeCN | 10 |
| 9 | KF (2.0) | DMA | 3 |
| 10 | CsF (2.0) | DMA | 16 |
| 11 | CsF (2.0) | DMF | 5 |
| 12 | CsF (2.0) | DMSO | 39 |
| 13 | CsF (2.0) | NMP | 63 |
| 14b | CsF (2.0) | NMP | 36 |
| 15c | CsF (2.0) | NMP | 76 |
| 16c | CsF (4.0) | NMP | 73 |
| 17 | CsF (4.0) | NMP | 52 |
| 18 | CsF (8.0) | NMP | 82 |
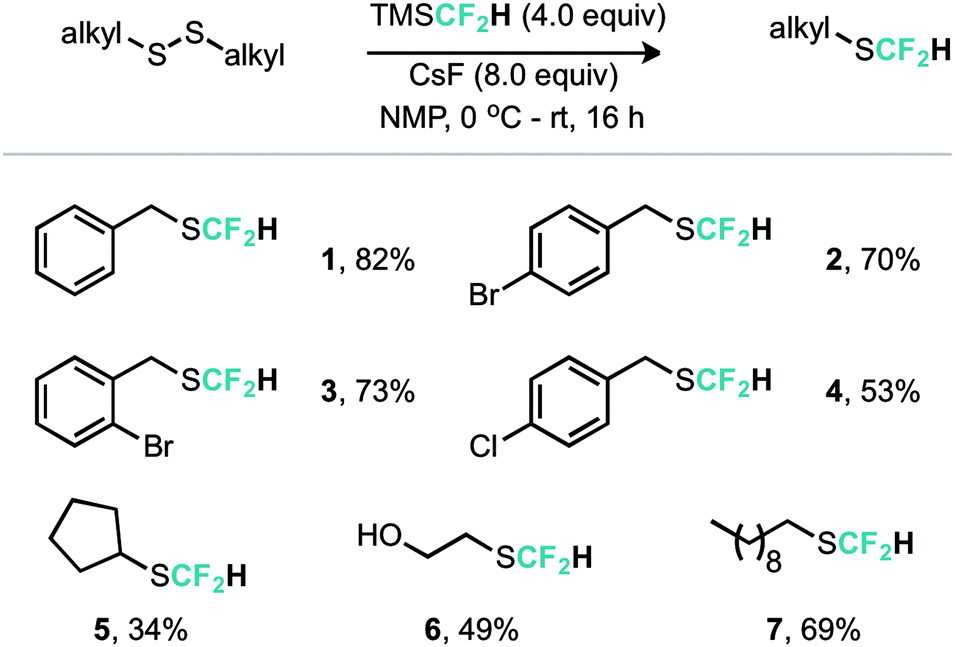 | ||
| Scheme 3 Reaction scope for dialkyldisulfides. | ||
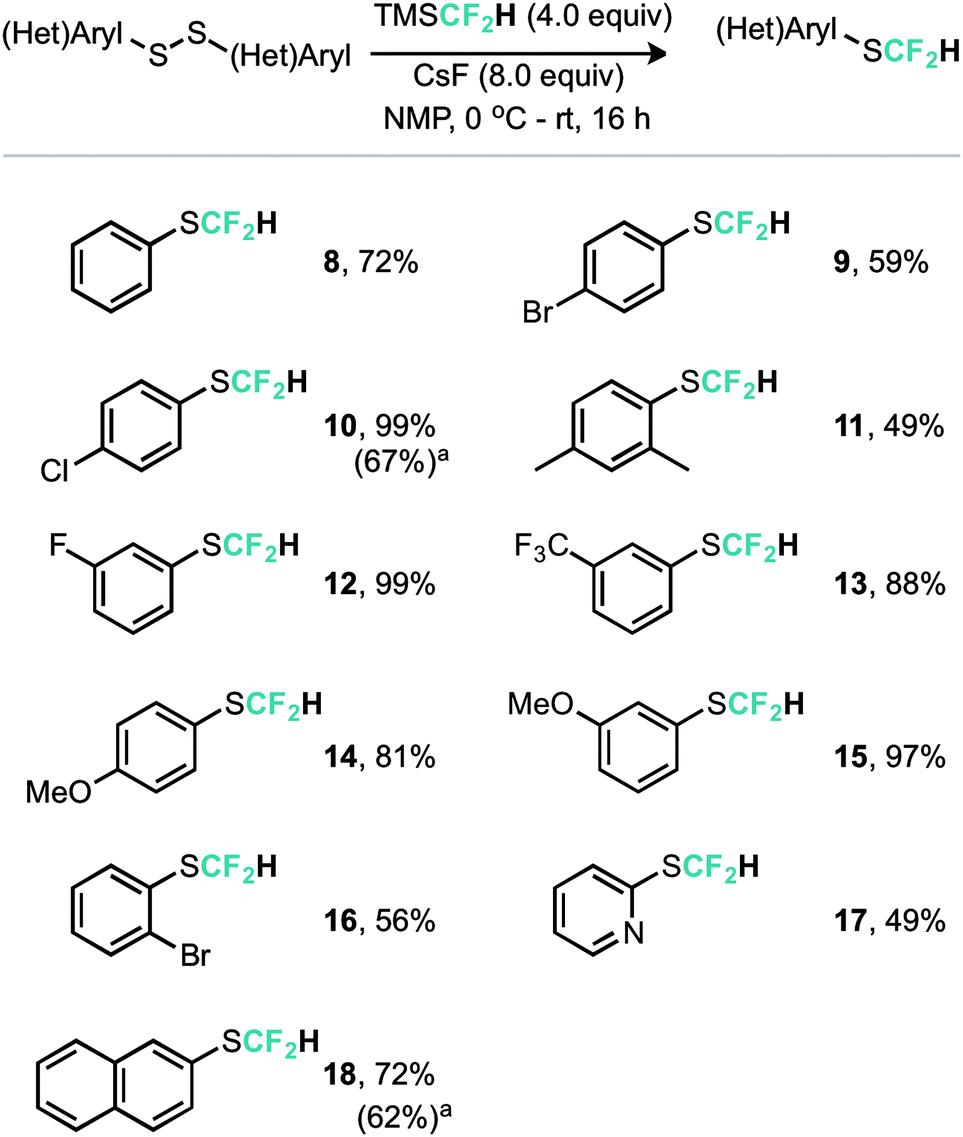 | ||
| Scheme 4 Reaction scope for diaryldisulfides. a Isolated yield.17 | ||
In summary, we report conditions for the preparation of a range of difluoromethylthioethers from their corresponding disulfide starting materials. The reported method is operationally simple, metal-free and uses commercially available fluorinating agents. The method is applicable to a range of dialkyldisulfides and diaryldisulfides and is tolerant to a range of functionalities, including free alcohols and pyridine nitrogens.
We thank the School of Chemistry, Cardiff University for generous support, Fluorochem for kind donations of TMSCF2H and the EPSRC UK National Mass Spectrometry Facility at Swansea University for mass spec data.
Notes and references
- For some overviews in the area of organofluorine chemistry see: (a) C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2015, 54, 3216 CrossRef CAS PubMed; (b) I. Hyohdoh, N. Furuichi, T. Aoki, Y. Itezono, H. Shirai, S. Ozawa, F. Watanabe, M. Matsushita, M. Sakaitani, P. S. Ho, K. Takanashi, N. Harada, Y. Tomii, K. Yoshinari, K. Ori, M. Tabo, Y. Aoki, N. Shimma and H. Iikura, ACS Med. Chem. Lett., 2013, 4, 1059 CrossRef CAS PubMed; (c) T. Furuya, C. A. Kuttruff and T. Ritter, Curr. Opin. Drug Discovery Dev., 2008, 11, 803 CAS; (d) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed; (e) J.-P. Bégué and D. Bonnet-Delpon, J. Fluorine Chem., 2006, 127, 992 CrossRef; (f) K. L. Kirk, Org. Process Res. Dev., 2008, 12, 305 CrossRef CAS; (g) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (h) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS PubMed; (i) N. A. Meanwell, J. Med. Chem., 2011, 54, 2529 CrossRef CAS PubMed; (j) D. T. Wong, K. W. Perry and F. P. Bymaster, Nat. Rev. Drug Discovery, 2005, 4, 764 CAS; (k) X.-H. Xu, K. Matsuzaki and N. Shibata, Chem. Rev., 2015, 115, 731 CrossRef CAS PubMed; (l) P. Jeschke, ChemBioChem, 2004, 5, 570 CrossRef CAS PubMed; (m) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (n) L. M. Yagupol'skii, A. Y. Il'chenko and N. V. Kondratenko, Russ. Chem. Rev., 1974, 43, 32 CrossRef; (o) D. L. Browne, Synlett, 2015, 33 CAS; (p) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS PubMed; (q) J.-A. Ma and D. Cahard, Chem. Rev., 2004, 104, 6119 CrossRef CAS PubMed.
- (a) B. E. Smart, J. Fluorine Chem., 2001, 109, 3 CrossRef CAS; (b) M. Morgenthaler, E. Schweizer, A. Hoffmann-Röder, F. Benini, R. E. Martin, G. Jaeschke, B. Wagner, H. Fischer, S. Bendels, D. Zimmerli, J. Schneider, F. Diederich, M. Kansy and K. Müller, ChemMedChem, 2007, 2, 1100 CrossRef CAS PubMed; (c) R. Filler and R. Saha, Future Med. Chem., 2009, 1, 777 CrossRef CAS PubMed; (d) D. B. Berkowitz and M. Bose, J. Fluorine Chem., 2001, 112, 13 CrossRef CAS; (e) D. O'Hagan, J. Fluorine Chem., 2010, 131, 1071 CrossRef.
- (a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (b) M. Schlosser, Angew. Chem., Int. Ed., 1998, 110, 1496 CrossRef; (c) A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS; (d) J. D. Dunitz and R. Taylor, Chem. – Eur. J., 1997, 3, 89 CrossRef CAS.
- (a) B. D. Key, R. D. Howell and C. S. Criddle, Environ. Sci. Technol., 1997, 31, 2445 CrossRef CAS; (b) D. O'Hagan, C. Schaffrath, S. L. Cobb, J. T. G. Hamilton and C. D. Murphy, Nature, 2002, 416, 279 CrossRef PubMed; (c) G. W. Gribble, Naturally Occurring Organofluorines, in Handbook of Environmental Chemistry, ed. A. H. Neilson, Springer-Verlag, Berlin, vol. 3, part N, 2002 Search PubMed.
- (a) X. Chen, S. Hussain, S. Parveen, S. Zhang, Y. Yang and C. Zhu, Curr. Med. Chem., 2012, 19, 3578 CrossRef CAS PubMed; (b) R. Bentley, Chem. Soc. Rev., 2005, 34, 609 RSC.
- (a) V. N. Boiko, Beilstein J. Org. Chem., 2010, 6, 880 CrossRef PubMed; (b) J. F. Giudicelli, C. Richer and A. Berdeaux, Br. J. Clin. Pharmacol., 1976, 3, 113 CrossRef CAS PubMed; (c) M. Diaferia, F. Veronesi, G. Morganti, L. Nisoli and D. P. Fioretti, Parasitol. Res., 2013, 112, 163 CrossRef PubMed.
- C. Hansch, A. Leo, S. H. Unger, K. H. Kim, D. Nikaitani and E. J. Lien, J. Med. Chem., 1973, 16, 1207 CrossRef CAS PubMed.
- (a) J. Hine and J. J. Porter, J. Am. Chem. Soc., 1960, 82, 6118 CrossRef CAS; (b) P. Deprez and J. P. Vevert, J. Fluorine Chem., 1996, 80, 159 CrossRef CAS; (c) Y. Zafrani, G. Sod-Moriah and Y. Segall, Tetrahedron, 2009, 65, 5278 CrossRef CAS; (d) K. Fuchibe, M. Bando, R. Takayama and J. Ichikawa, J. Fluorine Chem., 2015, 171, 133 CrossRef CAS; (e) V. P. Mehta and M. F. Greaney, Org. Lett., 2013, 15, 5036 CrossRef CAS PubMed; (f) G. K. Surya Prakash, S. Krishnamoorthy, S. Kar and G. A. Olah, J. Fluorine Chem., 2015, 180, 186 CrossRef; (g) W. Zhang, J.-M. Zhu and J.-B. Hu, Tetrahedron Lett., 2008, 49, 5006 CrossRef CAS; (h) Y. Fujiwara, J. A. Dixon, R. A. Rodriguez, R. D. Baxter, D. D. Dixon, M. R. Collin, D. G. Blackmond and P. S. Baran, J. Am. Chem. Soc., 2012, 134, 1494 CrossRef CAS PubMed; (i) W. Zhang, F. Wang and J. Hu, Org. Lett., 2009, 11, 2109 CrossRef CAS PubMed; (j) G. K. Surya Prakash, Z. Zhang, F. Wang, C.-F. Ni and G. A. Olah, J. Fluorine Chem., 2011, 132, 792 CrossRef; (k) L.-C. Li, F. Wang, C.-F. Ni and J.-B. Hu, Angew. Chem., Int. Ed., 2013, 52, 12390 CrossRef CAS PubMed.
- D. Zhu, Y. Gu, L. Lu and Q. Shen, J. Am. Chem. Soc., 2015, 137, 10547 CAS.
- J. Wu, Y. Gu, X. Leng and Q. Shen, Angew. Chem., Int. Ed., 2015, 54, 7648 CrossRef CAS PubMed.
- (a) B. Bayarmagnai, C. Matheis, K. Jouvin and L. J. Gooßen, Angew. Chem., Int. Ed., 2015, 54, 5753 CrossRef CAS PubMed; (b) K. Jouvin, C. Matheis and L. J. Gooßen, Chem. – Eur. J., 2015, 21, 14324 CrossRef CAS PubMed; (c) C. Matheis, M. Wang, T. Krause and L. J. Gooßen, Synlett, 2015, 1628 CAS; (d) G. Danoun, B. Bayarmagnai, M. F. Gruenberg and L. J. Gooßen, Chem. Sci., 2014, 5, 1312 RSC.
- During the preparation of this manuscript, we became aware of the work of J.-B. Han, H.-L. Qin, S.-H. Ye, L. Zhu and C.-P. Zhang, J. Org. Chem., 2016, 81, 2506 CrossRef CAS PubMed.
- T. Billard and B. R. Langlois, Tetrahedron Lett., 1996, 37, 6865 CrossRef CAS.
- T. Hagiwara and T. Fuchikami, Synlett, 1995, 717 CrossRef CAS.
- (a) Y. Zhao, W. Huang, J. Zheng and J. Hu, Org. Lett., 2011, 13, 5342 CrossRef CAS PubMed; (b) P. S. Fier and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 5524 CrossRef CAS PubMed.
- See the ESI† for details of this method adapted from A. Khazaei, M. A. Zolfigol and A. Rostami, Synthesis, 2004, 2959 CrossRef CAS.
- Isolated by a counter-current style extraction and column chromatography, see the ESI† for details. Value of ‘isolated yield’ proven to be largely subjective owing to the volatility of these compounds, 19F-NMR with an internal standard provides a less subjective measure of reaction performance.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc02693a |
| This journal is © The Royal Society of Chemistry 2016 |