
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence“Conformational lock” via unusual intramolecular C–F⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) C and C–H⋯Cl–C parallel dipoles observed in in situ cryocrystallized liquids†
C and C–H⋯Cl–C parallel dipoles observed in in situ cryocrystallized liquids†
Dhananjay
Dey
a,
Subhrajyoti
Bhandary
a,
Abhishek
Sirohiwal
a,
Venkatesha R.
Hathwar
b and
Deepak
Chopra
*a
aCrystallography and Crystal Chemistry Laboratory, Department of Chemistry, Indian Institute of Science Education and Research Bhopal, Bhopal By-Pass Road, Bhauri, Bhopal-462066, Madhya Pradesh, India. E-mail: dchopra@iiserb.ac.in; Fax: +91 755 6692392
bDepartment of Chemistry and iNANO Aarrhus University, Langelandsgade 140, Aarhus C DK-8000, Denmark
First published on 18th April 2016
Abstract
We report an unusual intramolecular C–F⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–H⋯Cl–C parallel dipole–dipole alignment which “locks” the molecular conformation of cryocrystallized liquids towards planarity where the diatropic ring current establishes the existence of aromaticity in the five-membered ring associated with F⋯O contact. Topological analysis establishes the bonding interaction between [F, O] and [H, Cl].
C and C–H⋯Cl–C parallel dipole–dipole alignment which “locks” the molecular conformation of cryocrystallized liquids towards planarity where the diatropic ring current establishes the existence of aromaticity in the five-membered ring associated with F⋯O contact. Topological analysis establishes the bonding interaction between [F, O] and [H, Cl].
Non-covalent interactions have an enormously significant role in chemical as well as biological systems,1,2 especially intramolecular non-covalent interactions that define the stability and the molecular conformation of the compound.3 The increase in the number of fluorinating agents demand the detail investigation of intramolecular interactions for modern structure-based design strategies in organofluoro compounds.4 Due to the high electronegativity and lesser polarizability of the fluorine atom, the existence of a possible interaction between F and O is deemed questionable. However, the scope for the existence of an intuitive F⋯O interaction will exist only when the carbon bonded fluorine atom is in an environment connected to sufficient electron withdrawing group/groups.5 The anisotropic electron distribution around the halogen atom leads to a decrease in the repulsion and an increase in the electrostatic attraction in X⋯N/O.6 According to Allen et al.,6 based on the CSD study, the interaction between F and related electronegative atoms does not occur at all. The strength of the investigated interactions is strongly dependent on the geometrical parameters and the underlying concept of the existence of the σ-hole.7 Intramolecular non-covalent interaction (O⋯S, N⋯S and F⋯S8) has an important role in overall molecular conformation towards planarity in conjugated polymers and small molecules.9 Furthermore, an intramolecular S⋯O interaction, reported previously determines the stereoselectivity in an asymmetric Pummerer reaction.10
The existence of halogen bonding involving the heavier halogen atoms Cl/Br/I⋯N/O (intramolecular)11 is well known in the literature. To date, however, there is no experimental evidence in favor of the existence of a possible short C–F⋯OC contact. In the current study, we have used the method of in situ cryocrystallization and observed the possibility of parallel alignment of C–F and OC; C–Cl and C–H bond dipoles in the crystal structures of polyfluorinated benzoyl chlorides12–14 (Scheme 1). The cryocrystallization of the parent compound i.e. benzoyl chloride (BC), (Fig. S1–S11 and Tables S1 and S2, ESI†) has also been performed for a comparison of the electronic features in the absence of the C–F bond. The most notable feature is the observation of a short intramolecular contact between the fluorine and the oxygen atom which is observed in the case of ortho fluoro substituted benzoyl chloride and related compounds in the presence of intramolecular short C–H⋯Cl contact.
 | ||
| Scheme 1 Nomenclature of fluorinated benzoyl chlorides. | ||
To investigate into the nature and energetics of these contacts, a detailed investigation of the planar geometry was performed. This was further supported via detailed input from QTAIM,15,16 distributed atomic polarizability,17 NICS18,19 method and the NCI descriptor20–22 to characterize these contacts in the solid state (Computational methodologies in ESI†).
Interestingly, in the molecular conformation of 2-fluoro benzoyl chloride (2FBC) the torsion angle between the benzoyl group and the fluoro phenyl ring is 5° in the solid state and this value is 0° for the optimized geometry (Fig. 1). Furthermore, in the solid-state, the trends in molecular conformation for different cryocrystallized liquids are in the range of 3.5–17.0° (Fig. S12 and Table S3, ESI†). Only in case of 26DFBC is the –COCl group more relaxed (53.0°) when compared to the solid state geometry (44.1°) for both the symmetry independent molecules (Fig. 1). In the solid state geometry, the distance between the fluorine and oxygen atom is in the range of 2.637 to 2.776 Å (inter-nuclear distance) whereas the distances for H⋯Cl contact are 2.62 Å and 2.54 Å, and the directionalities are 104° and 106° in case of BC and 2FBC respectively.
 | ||
| Fig. 1 Change in molecular conformation from the solid state to the gas phase geometry for 2FBC and 26DFBC_1 respectively. | ||
In order to understand comprehensively the role of these contacts in “conformational lock” and overall stability, a conformational analysis has been performed for 2FBC and 26DFBC_1. The relative potential energy vs. the dihedral angle plot around the C–C bond connecting the –COCl group with respect to the aryl ring shows that the nature of the plot for 2FBC is opposite to that observed in the case of 26DFBC_1 (Fig. 2). In 2FBC, the conformation corresponding to the minimum potential energy leads to the –COCl group and the phenyl ring in the same plane whereas in the case of 26DFBC_1, the molecular geometry does not correspond to a minimum. The relative energy difference between the planar conformation (0°) and the –COCl group aligned normal to the ring plane (90°) is 4.38 kcal mol−1 in case of 2FBC and 6.5 kcal mol−1 in case of BC. This is indeed noteworthy on account of the fact that the resonance assisted delocalization of electron density from fluorine to the benzoyl group decreases the electrophilicity of the carbonyl carbon. This in turn reduces the overall bond dipole moment for C–Cl bond resulting in overall reduced stability for the molecular conformation in case of 2FBC when compared with BC. This is also reflected in the fact that for a conformational change of 20°, the energy change for 2FBC is lower in comparison to 2BC. Thus, the analysis clearly reveals the stabilizing role of the C–F⋯OC contact in the presence of intramolecular C–H⋯Cl contact to adopt the existing geometry.
 | ||
| Fig. 2 Plot of relative potential energy vs. dihedral angle for BC, 2FBC and 26DFBC_1. | ||
To understand further the nature of the intramolecular F⋯O and H⋯Cl contact, a detailed QTAIM analysis was performed to obtain the topological parameters for all fluorinated benzoyl chlorides (Fig. 3 and Fig. S14, ESI†). Except for 26DFBC, other ortho fluoro substituted benzoyl chlorides show the presence of a (3, −1) bond critical point (BCP, shown in a brown colored circle) for the C–F⋯OC contact and a ring critical point (3, +1) [shown in a red colored circle] of the five-membered ring. The BCP and RCP were also observed for the intramolecular H⋯Cl contact in BC and 2FBC. The topological parameters for Cl⋯F and H⋯Cl are given in Table S5 (ESI†). In the case of 26DFBC, the F⋯O BCP is absent. On the contrary, a Cl⋯F BCP exists. The value of the electron density (ρBCP) and Laplacian (∇2ρBCP) at the BCP for F⋯O contact and H⋯Cl are in the range of 0.093–0.101 e Å−3 and 1.449–1.554 e Å−5; 0.094–0.106 e Å−3 and 1.378–1.512 e Å−5 respectively (Table 1 and Table S5, ESI†). The corresponding bond dissociation energies23 are in the range of 3.46–4.14 kcal mol−1 for F⋯O contact and 3.00–3.39 kcal mol−1 for H⋯Cl contact, respectively. These values are comparable with a strong hydrogen bond.24,25 Study of the Cambridge Structural Database (CSD) reveals that the number of organic molecules with similar intramolecular F⋯O contacts is 10, in which the BCP between F and O atom was observed (Fig. S13 and Table S4, ESI†) in the absence of any other intramolecular contact.
 | ||
| Fig. 3 Molecular graphs for (a) BC, (b) 2FBC, (c) 23DFBC, (d) 24DFBC, (e) 25DFBC_1, (f) 25DFBC_2 depicting bond critical points at intramolecular Cl⋯H and F⋯O bond paths. (g) 26DFBC_1 and (h) 26DFBC_2 wherein the Cl⋯F BCP and RCP are shown. | ||
C contact
| Code | R ij/Å | ρ BCP/e Å−3 | ∇2ρBCP/e Å−5 | V b (a.u.) | G b (a.u.) | DEV/kcal mol−1 |
|---|---|---|---|---|---|---|
| a Optimized geometry. b Theoretical multipole refinement. | ||||||
| 2FBC | 2.64 | 0.101 | 1.554 | −0.013184 | 0.014649 | 4.14 |
| 2FBC | 2.66 | 0.096 | 1.462 | −0.012480 | 0.013820 | 3.92 |
| 2FBC | 2.64 | 0.093 | 1.785 | −0.010701 | 0.014594 | 3.36 |
| 23DFBC | 2.66 | 0.097 | 1.480 | −0.012591 | 0.013968 | 3.95 |
| 24DFBC | 2.67 | 0.095 | 1.468 | −0.012391 | 0.013809 | 3.89 |
| 25DFBC_1 | 2.68 | 0.094 | 1.449 | −0.012282 | 0.013654 | 3.85 |
| 25DFBC_2 | 2.65 | 0.098 | 1.514 | −0.012808 | 0.014243 | 4.02 |
| BOMRIG | 2.75 | 0.083 | 1.334 | −0.011043 | 0.012162 | 3.46 |
| COTPUX | 2.70 | 0.091 | 1.317 | −0.011943 | 0.012801 | 3.75 |
| EVAJIV | 2.69 | 0.094 | 1.411 | −0.012325 | 0.012325 | 3.89 |
| GAPJIS | 2.76 | 0.085 | 1.332 | −0.011291 | 0.012554 | 3.54 |
| HAWXEJ | 2.70 | 0.090 | 1.351 | −0.011556 | 0.012784 | 3.63 |
| HICFEE | 2.69 | 0.092 | 1.373 | −0.011697 | 0.012967 | 3.67 |
Theoretical multipole charge density refinement (from XD) on the experimentally observed geometry (Fig. S15–S17, ESI†) of 2FBC reveals that the electron density and the Laplacian at the BCP for the intramolecular C–F⋯OC contact are 0.093 e Å−3 and 1.758 e Å−5; the values are 0.102 e Å−3 and 1.612 e Å−5 at the BCP for Cl⋯H contact, establishing the closed-shell nature of these contacts and qualifies this is a “bonding interaction”. Fig. 4 shows the static 3D deformation density map for 2FBC obtained from multipole refinements. The charges, from the integration of the atomic basin, on the carbon atoms attached to –COCl and –F group are 0.63 and 0.44e, respectively, thereby indicating the charge deficient regions (in red) on these atoms which support the charge concentration regions (in blue) present on the electronegative atoms fluorine and oxygen.
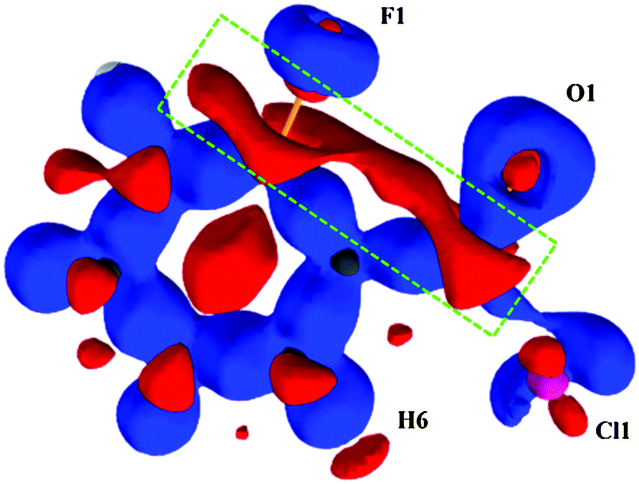 | ||
| Fig. 4 3D deformation density map obtained from the theoretical multipole model for 2FBC. The blue regions indicate charge concentrated (CC) regions and red regions indicate charge depleted (CD) regions. The contour intervals are at ±0.10 e Å−3. | ||
To investigate, the “lock” mechanism an understanding of the influence of the –COCl group on the vicinal atoms (F1, H2 and H6) in terms of distributed atomic polarizability in BC and 2FBC will be helpful. The distributed atomic polarizability has been calculated using QTAIM based topological partitioning of the electron density for BC and 2FBC using PolaBer.26 In Fig. 5, we have compared the atomic polarizabilities of BC and 2FBC (Table S6, ESI†). The polarizability ellipsoid of the oxygen atom in BC is elongated along the covalent bond, however in 2FBC, the inclusion of the fluorine atom alters the direction and the volume of the ellipsoid. In addition the polarizability ellipsoid of oxygen in 2FBC is more anisotropic when compared to BC (αiso & VA in Table S6, ESI†). Large deviations were also noticed in the polarizability tensor component of oxygen in BC compared to the oxygen of 2FBC; for example, α22 in BC was found to be 3.668 a.u., whereas in 2FBC it is 9.905 a.u. Ellipsoid axes depict the anisotropy of the atomic polarizability, thus in 2FBC, atomic electron density of oxygen is more polarized towards fluorine which operates “through space”. It was also observed that the atomic electron density of H6 is more polarizable (αiso for H2 and H6 are 1.930 a.u. and 2.488 a.u. in BC; αiso for H6 is 2.422 a.u. in 2FBC) than that of H2 (not involved in intramolecular contact) in BC due to H⋯Cl contact. As a consequence, the polarizability ellipsoid of H6 in BC and 2FBC is oriented towards the direction of the H⋯Cl contact. This can also be justified from the atomic charges (obtained from XD) on fluorine and oxygen being −0.74e and −1.23e, respectively (wherein, the atomic charges for Cl and H are −0.44e and 0.08e respectively), and hence the effect of the former atom on the latter.
 | ||
| Fig. 5 Distributed atomic polarizability ellipsoids for (a) BC and (b) 2FBC. Intramolecular Cl⋯H, F⋯O contact is shown as a dashed line. The scaling factor for ellipsoids is 0.3 Å−2. (Note: atomic polarizability tensors are depicted as ellipsoids, visualization is done in same real space as the molecule assuming 1 Å3 ≡ 1 Å, thus a scaling factor is used for better visualization of the ellipsoids). | ||
Furthermore, to characterize the aromaticity of the five-membered ring associated with intramolecular C–F⋯OC contact, nucleus-independent chemical shift (NICS) analysis has been performed up to a 2 Å distance from the ring critical point (RCP) along the normal to the ring plane with an interval of 0.1 Å. NICS(0)zz. The value of the diatropic ring current for σ + π-electron delocalization at the RCP is −17.82 ppm (Fig. 6), whereas the diatropic ring current only for π-electron delocalization NICS(1)zz value at 1 Å above the ring plane is −3.83 ppm, indicating the presence of aromaticity due to the phenomenon of resonance delocalization, the planar geometry being a pre-requisite for the same. In addition to this, the delocalization index27 [δ(F, O)] for C–F⋯OC contact was found to be 0.0614e ([δ(H, Cl)] is 0.0571e), which indicates that the average number of delocalized electrons between two interacting atoms (from AIMALL) is comparable to those of intermolecular H-bonds.28 Furthermore, we have investigated the NCI region surrounding the BCP in terms of the reduced density gradient (RDG) isosurfaces in real space. The sign of the second eigenvalue (λ2) of the Hessian matrix distinguishes whether the intramolecular C–F⋯OC and C–H⋯Cl–C contact are stabilized (λ2 < 0) or destabilized (λ2 > 0) (Fig. S18–S25 and Table S7, ESI†). The RDG isosurfaces for fluorinated benzoyl chlorides are shown in Fig. 7 with the color range of −0.03 < ρ * sign (λ2) < 0.03 a.u.
 | ||
| Fig. 6 NICS values as a function of distance (Å) of the five-membered ring associated with intramolecular F⋯O contact in 2FBC. | ||
 | ||
| Fig. 7 RDG isosurface for intramolecular F⋯O, H⋯Cl and Cl⋯F contacts with s = 0.6. The color is plotted in the range of −0.03 < ρ * sign (λ2) < 0.03 a.u. | ||
The analysis reveals the NCI trough (around the bond critical point) is a combination of a green RDG isosurface with negative eigenvalue (λ2 < 0) corresponding to a weak interaction between (F, O) and (H, Cl) atom, and a blue RDG isosurface (λ2 > 0) corresponding to the ring critical point (Fig. 7). The plot of RDG vs. sign (λ2) * ρ for 2FBC gives one spike in the negative region with a value of −0.02 a.u. further supporting the existence of dispersive vdW interaction (Fig. S26, ESI†) that stabilized the planar molecular conformation with a minimum C–F⋯OC distance and a short Cl⋯H contact.
To summarize, we report here the first crystallographic evidence of an intramolecular C–F⋯OC contact observed in fluoro-substituted benzoyl chlorides which are a very common precursor in organic synthesis. The aim of this study was to investigate and characterize the existence of this contact in the presence of C–H⋯Cl contact, which facilitates a “conformation locking” mechanism along with global stabilization via the existence of the planar conformation. It is well supported by QTAIM analysis, theoretical multipole refinement, distributed atomic polarizability calculations, NICS and NCI-RDG analysis around the (3, −1) BCP which establishes it as a “bonding interaction”. Atomic electron density of the oxygen atom in 2FBC is more polarized towards F⋯O contact which is very well supported by the NCI approach, which establishes the dispersive nature responsible for the stabilization of the molecular conformation in the solid state. Hence, this study is significant, keeping in mind that dipole–dipole interactions govern the reactivity of organic molecules and this factor plays an important role in the elucidation of the mechanisms for certain classes of organic reactions. Futuristic studies shall aim towards the investigation of the effect of different electron donating and withdrawing substituents on the nature of the C–F⋯OC contact.
D. D. and S. B. thank IISERB for the research fellowship. D. C. and D. D. thank IISERB for the research facilities, infrastructure, and DST-SERB for research funding. We would like to thank Prof. R. Boese and Dr A. R. Choudhury for valuable discussions and Dr Gabriele Saleh for providing inputs for NCImilano. We would also like to thank Dr Piero Macchi & Dr Anna Krawczuk for invaluable inputs in atomic polarizability calculations.
Notes and references
- J. F. Xu, L. Chen and X. Zhang, Chem. – Eur. J., 2015, 21, 11938 CrossRef CAS PubMed.
- H. Y. Yu, T. Ziegelhoffer, J. Osipiuk, S. J. Ciesielski, M. Baranowski, M. Zhou, A. Joachimiak and E. A. Craig, J. Mol. Biol., 2015, 427, 1632 CrossRef CAS PubMed.
- A. J. Mukherjee, S. S. Zade, H. B. Singh and R. B. Sunoj, Chem. Rev., 2010, 110, 4357 CrossRef CAS PubMed.
- G. T. Giuffredi, V. Gouverneur and B. Bernet, Angew. Chem., Int. Ed., 2013, 52, 10524 CrossRef CAS PubMed.
- D. Chopra, Cryst. Growth Des., 2012, 12, 541 CAS.
- J. P. M. Lommerse, A. J. Stone, R. Taylor and F. H. Allen, J. Am. Chem. Soc., 1996, 118, 3108–3116 CrossRef CAS.
- P. Metrangolo and G. Resnati, Cryst. Growth Des., 2012, 12, 5835 CAS.
- M. S. Pavan, K. D. Prasad and T. N. G. Row, Chem. Commun., 2013, 49, 7558 RSC.
- N. E. Jackson, B. M. Savoie, K. L. Kohlstedt, M. O. de la Cruz, G. C. Schatz, L. X. Chen and M. A. Ratner, J. Am. Chem. Soc., 2013, 135, 10475 CrossRef CAS PubMed.
- Y. Nagao, S. Miyamoto, M. Miyamoto, H. Takeshige, K. Hayashi, S. Sano, M. Shiro, K. Yamaguchi and Y. Sei, J. Am. Chem. Soc., 2006, 128, 9722 CrossRef CAS PubMed.
- D. L. Widner, Q. R. Knauf, M. T. Merucci, T. R. Fritz, J. S. Sauer, E. D. Speetzen, E. Bosch and N. P. Bowling, J. Org. Chem., 2014, 79, 6269 CrossRef CAS PubMed.
- A. R. Choudhury, N. Winterton, A. Steiner, A. I. Cooper and K. A. Johnson, J. Am. Chem. Soc., 2005, 127, 16792 CrossRef CAS PubMed.
- R. Boese, D. Blaser and G. Jansen, J. Am. Chem. Soc., 2009, 131, 2104 CrossRef CAS PubMed.
- R. Boese, M. T. Kirchner, W. E. Billiups and L. R. Norman, Angew. Chem., Int. Ed., 2003, 42, 1961 CrossRef CAS PubMed.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford, U.K., 1990 Search PubMed.
- V. G. Tsirelson, in The Quantum Theory of Atoms in Molecules: From Solid State to DNA and Drug Design, ed. C. Matta and R. Boyd, Wiley-VCH, Weinheim, Germany, 2007, ch. 10, p. 45 Search PubMed.
- A. Krawczuk-Pantula, D. Pérez, K. Stadnicka and P. Macchi, Trans. Am. Crystallogr. Assoc., 2011, 42, 1 Search PubMed.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. V. R. Schleyer, Chem. Rev., 2005, 105, 3842 CrossRef CAS PubMed.
- P. V. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. V. E. Hommes, J. Am. Chem. Soc., 1996, 118, 6317 CrossRef CAS.
- G. Saleh, L. L. Presti, C. Gatti and D. Ceresoli, J. Appl. Crystallogr., 2013, 46, 1513 CAS.
- G. Saleh, C. Gatti, L. L. Presti and J. Contreras-Garc, Chem. – Eur. J., 2012, 18, 15523 CrossRef CAS PubMed.
- J. Contreras-Garcia, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625 CrossRef CAS PubMed.
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170 CrossRef CAS.
- D. Dey, T. P. Mohan, B. Vishalakshi and D. Chopra, Cryst. Growth Des., 2014, 14, 5881 CAS.
- D. Dey, S. P. Thomas, M. A. Spackman and D. Chopra, Chem. Commun., 2016, 52, 2141 RSC.
- A. Krawczuk, D. Perez and P. Macchi, J. Appl. Crystallogr., 2014, 47, 1452 CAS.
- R. F. W. Bader, A. Streitwieser, A. Neuhaus, K. E. Laidig and P. Speers, J. Am. Chem. Soc., 1996, 118, 4959 CrossRef CAS.
- L. Guillaumes, P. Salvador and S. Simon, J. Phys. Chem. A, 2014, 118, 1142 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Thermal characterization; in situ crystallization procedure (crystal growth and SCXRD data collections); CSD search; and various computational methodologies. CCDC 1440618, 1440619, 1452040 and 1426052–1426054. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc02535h |
| This journal is © The Royal Society of Chemistry 2016 |