
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRadical perfluoroalkylation – easy access to 2-perfluoroalkylindol-3-imines via electron catalysis†
Dirk
Leifert
,
Denis G.
Artiukhin
,
Johannes
Neugebauer
,
Anzhela
Galstyan
,
Cristian Alejandro
Strassert
and
Armido
Studer
*
Westfälische Wilhelms-Universität Münster, Corrensstraße 40, 48149 Münster, Germany. E-mail: studer@uni-muenster.de
First published on 31st March 2016
Abstract
Arylisonitriles (2 equivalents) react with alkyl and perfluoroalkyl radicals to form 2-alkylated indole-3-imines via two sequential additions to the isonitrile moiety followed by homolytic aromatic substitution. The three component reaction comprises three C–C bond formations. The endocyclic imine functionality in the products is more reactive in follow up chemistry and hydrolysis of the exocyclic imine leads to 3-oxindoles that show fluorescence properties.
Indoles generally show high biological activity1 and accordingly they are observed to be prominent substructures in many natural products and drug candidates.2 Therefore, the development of novel methods allowing easy access to indoles and their derivatives is of importance.3 The introduction of a CF3 group into a lead compound has become a general strategy to further improve lipophilicity, bioactivity and metabolic stability4 of the given lead candidate in agrochemical and medicinal chemistry.5 Along these lines, the synthesis of 2-trifluoromethylindoles has gained great attention recently.6
Radical chemistry has become valuable for simple and efficient incorporation of CF3 groups into various compounds via direct trifluoromethylation.7 Recently, we8 and Yu et al.9 introduced the radical trifluoromethylation of 2-isocyanobiphenyls as a practical method for the modular synthesis of 6-trifluoromethylated phenanthridines.10,11 Isonitriles have also been successfully applied to the preparation of 2-trifluoromethylated indoles 3via radical trifluoromethylation of 2-isocyanostyrene derivatives 1 with the Togni reagent 2a (Fig. 1a).12,13
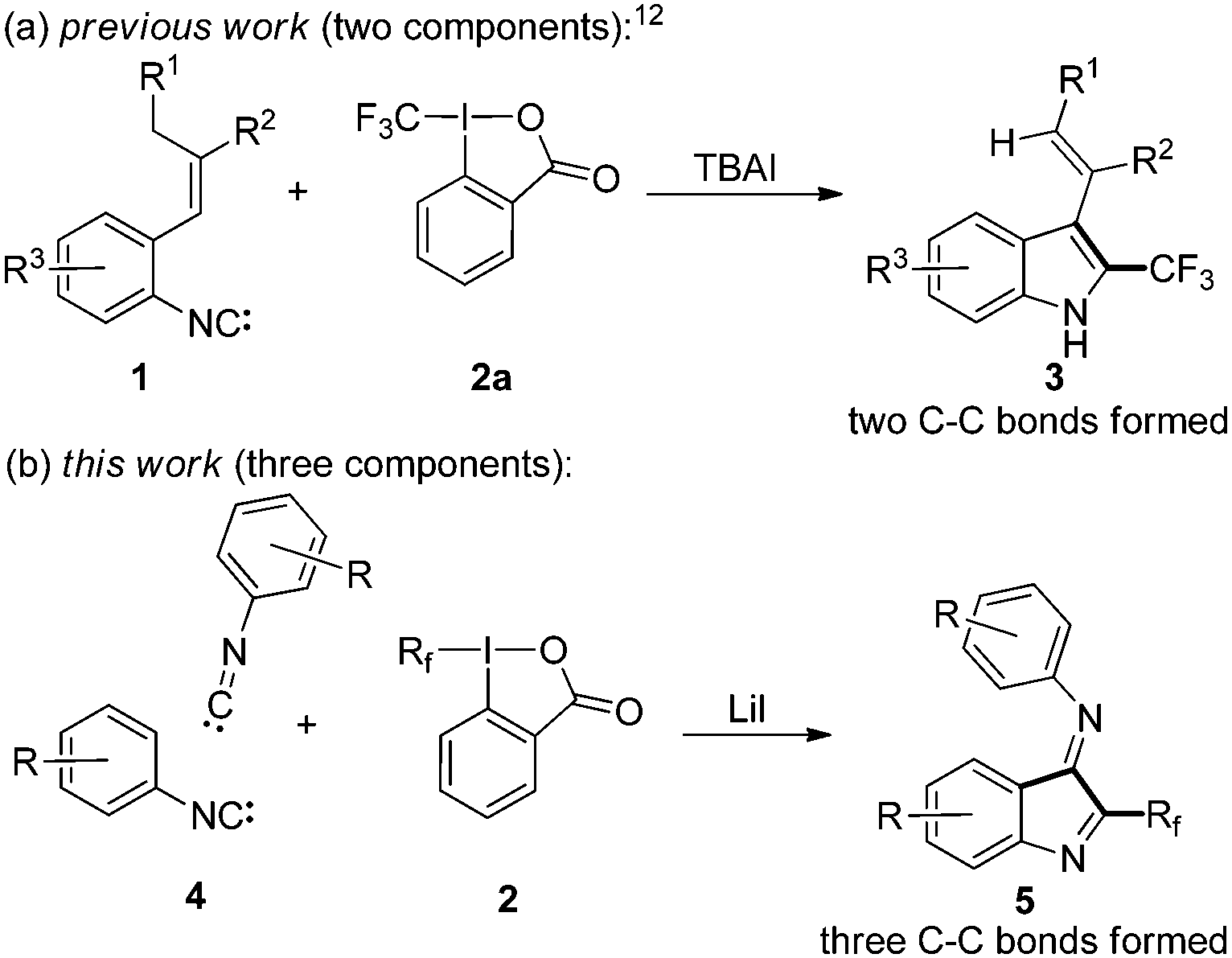 | ||
| Fig. 1 Strategies towards 2-perfluoroalkyl indoles. | ||
The advantages of these radical trifluoromethylation methods over existing processes6,14 are that transition metals are not required and the introduction of the CF3 group occurs regioselectively. Moreover, radical chemistry is ideally suited to run cascades allowing for multiple C–C bond formations. Herein we report the preparation of 2-perfluoroalkylindol-3-imines 5via a three component cascade comprising two subsequent radical additions to isonitriles 4 using reagents of type 2 (Fig. 1b).15
4-Methoxyphenylisonitrile (4a) was used as a test substrate for optimization (Table 1). Pleasingly, the reaction of 4a (4.8 equiv.), 2a (1.0 equiv.) as a CF3 source and tetrabutylammonium iodide (TBAI, 4.8 mol%) as an initiator at 80 °C in 1,4-dioxane for 22 hours provided 40% of the targeted 2-trifluoromethylindol-3-imine 5a (Table 1, entry 1). Solvent screening revealed that ethyl acetate among the tested solvents provides the highest yield (Table 1, entries 2–4). We also varied the amount of isonitrile 4a and found that with 3.8 equiv. the best result is obtained. 90 °C was identified as the ideal reaction temperature (Table 1, entries 6, 8 and 9) and 4.8 mol% of TBAI-initiator was sufficient (54%). Reactions at higher (0.52 molar) or at lower (0.09 molar) concentration provided lower yields (see the ESI,† Table S1). Among the iodide sources tested (LiI, NaI, KI, CsI, MgI2, CaI2), LiI provided the highest yield (60%) (Table 1, entries 8 and 12 and ESI,† Table S1). Addition of an external base (Li2CO3 or LiOH) did not lead to any further improvement of the yield (Table 1, entries 13 and 14).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Equiv. (4a) | Init. (mol%) | Solvent | Temp. (°C) | Yielda (%) |
| a Isolated yield (reactions run at 0.26 molar). b Addition of Li2CO3 (0.25 mmol). c Addition of LiOH (0.25 mmol). | |||||
| 1 | 4.8 | TBAI (4.8) | Dioxane | 80 | 40 |
| 2 | 4.8 | TBAI (4.8) | CHCl3 | 80 | 40 |
| 3 | 4.8 | TBAI (4.8) | MeCN | 80 | 35 |
| 4 | 4.8 | TBAI (4.8) | EtOAc | 80 | 41 |
| 5 | 2.9 | TBAI (4.8) | EtOAc | 80 | 36 |
| 6 | 3.8 | TBAI (4.8) | EtOAc | 80 | 41 |
| 7 | 9.6 | TBAI (4.8) | EtOAc | 80 | 19 |
| 8 | 3.8 | TBAI (4.8) | EtOAc | 90 | 54 |
| 9 | 3.8 | TBAI (4.8) | EtOAc | 100 | 47 |
| 10 | 3.8 | TBAI (2.9) | EtOAc | 90 | 54 |
| 11 | 3.8 | TBAI (9.6) | EtOAc | 90 | 42 |
| 12 | 3.8 | LiI (4.9) | EtOAc | 90 | 60 |
| 13 | 3.8 | LiI (4.9) | EtOAc | 90 | 57b |
| 14 | 3.8 | LiI (4.9) | EtOAc | 90 | 58c |
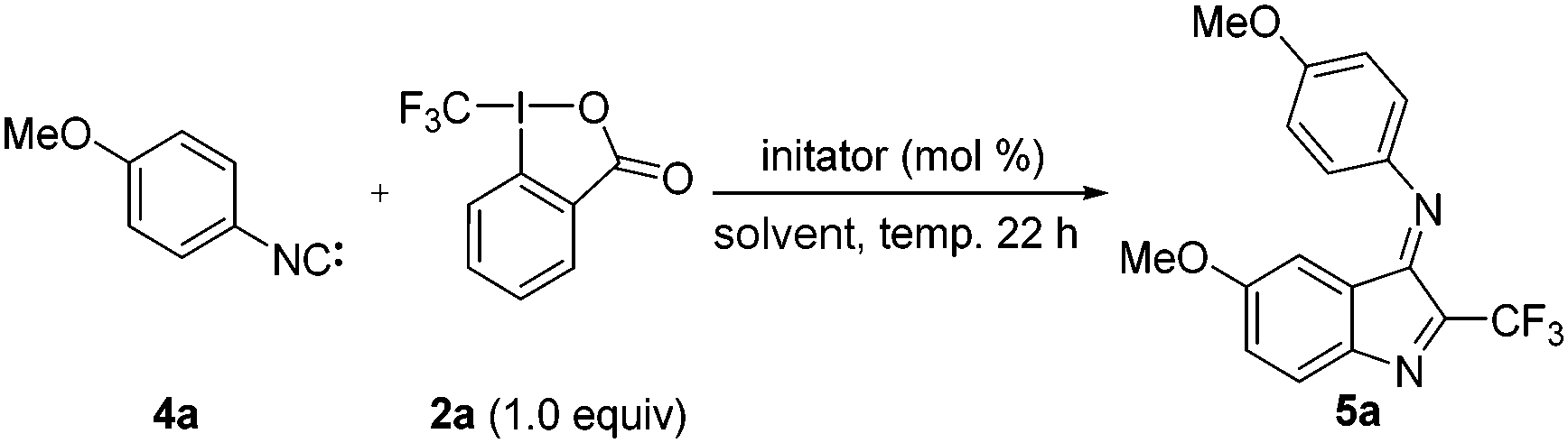
Under optimized conditions (Table 1, entry 13) we next investigated the scope and limitations (Fig. 2). Electronic effects strongly influence the reaction outcome and generally better results are obtained for isonitriles bearing electron-donating groups at the para-position. The silyloxy-isonitrile 4b in the reaction with 2a provided 5b in 41% and the dimethylamino-derivative 4d gave 5d in 39% yield. Alkyl-substituted phenyl isonitriles 4e and 4f provided the methyl and the tert-butyl derivatives 5e and 5f in 32% and 22% yields. The yield further dropped for the methylthiyl-isonitrile and the parent phenylisonitrile (see 5c, 5g). The lower yields for the electron deficient systems are likely caused by the low stability of the corresponding isonitriles.16 Reaction with the m-MeOC6H4CN 4h provided a mixture of the two regioisomers 5h and 5h′ with a 2.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 selectivity. The selectivity was far higher (8:1) for 5-isocyanobenzo[d][1,3]dioxole 4i to give 5i as the major isomer.
:1 selectivity. The selectivity was far higher (8:1) for 5-isocyanobenzo[d][1,3]dioxole 4i to give 5i as the major isomer.
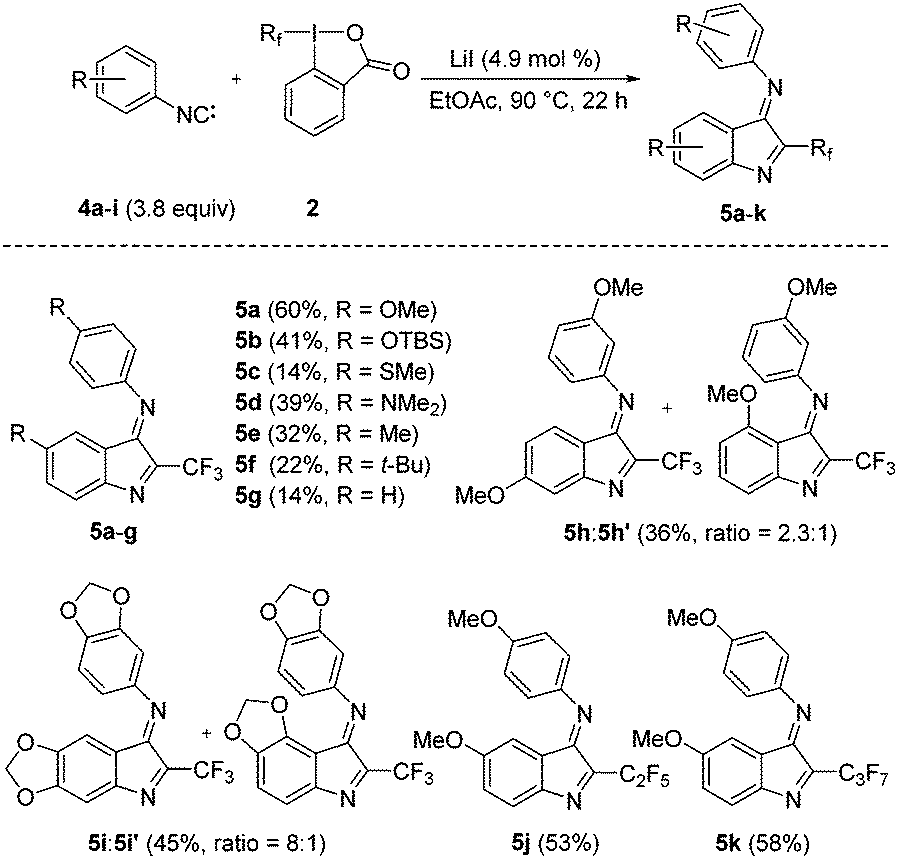 | ||
| Fig. 2 Various prepared 2-trifluoromethyl or perfluoroalkylindol-3-imines 5a–k (isolated yields). | ||
Homologues of Togni reagent 2a bearing longer perfluoroalkyl chains (C2F5 and C3F7) in the reaction with 4a gave the indoles 5j and 5k in 53% and 58% yields, respectively.
The suggested mechanism comprising a base-promoted homolytic aromatic substitution17via electron-catalysis18 is depicted in Fig. 3. The initiation of the cascade likely occurs by reduction of the I(III)-reagent 2 with LiI as a formal electron donor8,19 to generate a CF3 radical. It is likely that there is an initial substitution of the carboxylate functionality of 2 by the iodide to give an aryl-I(III)ICF3-compound. The I–I-bond is weak and upon homolysis the generated iodanyl radical fragments to give a CF3 radical and 2-iodobenzoate. This CF3-radical adds to 4a to give the imidoyl radical A, which further adds to a second isonitrile to generate the imidoyl radical B. Cyclization onto the arene of the first aryl isonitrile leads the cyclohexadienyl radical C, that gets deprotonated by 2-iodobenzoate8 to form the radical anion D. 2-Iodobenzoate is generated in the chain by reduction of 2. D is an efficient SET reducing reagent which formally liberates an electron giving product 5a thereby closing the catalytic cycle.17
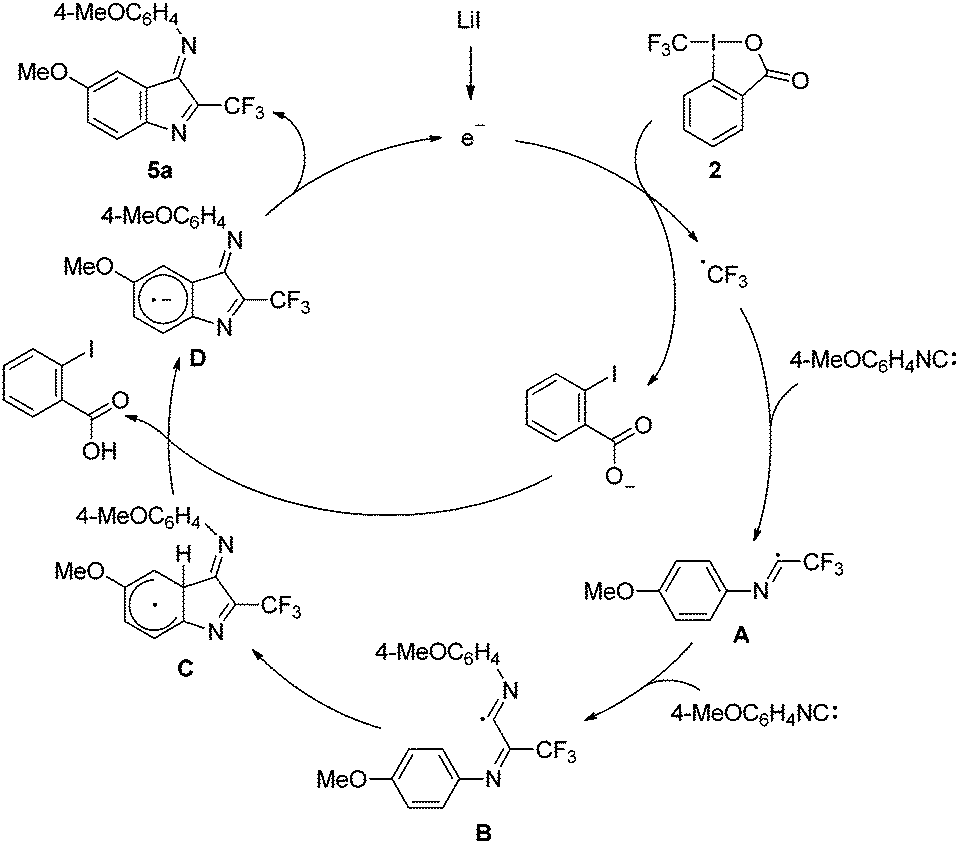 | ||
| Fig. 3 Suggested mechanism exemplified by the synthesis of 5a. | ||
Due to the high values of these indole imines (see below) we decided to develop an alternative non-chain process to the target compounds that uses readily accessible azo compounds as radical precursors (Fig. 4).11b After some experimentation we found that heating of isonitrile 4a (3.8 equiv.) in the presence of AIBN (azobis(isobutyronitrile)) 6a in benzene at 100 °C provided the 2-cyanoprop-2-yl indole imine 7a in 81% yield. The yield is calculated on the basis that one equivalent of AIBN is necessary for product formation (AIBN acts as a radical precursor and an oxidant)20 and that only about 60% of the AIBN-derived radicals escape the solvent cage.21 In analogy, by using 1,1′-azobis(cyclohexanecarbonitrile) 6b the indole 7b was obtained in 78% yield. The yield further improved upon switching to the ester derivative 6c as a radical precursor to give the indole 7c in a high yield (92%).
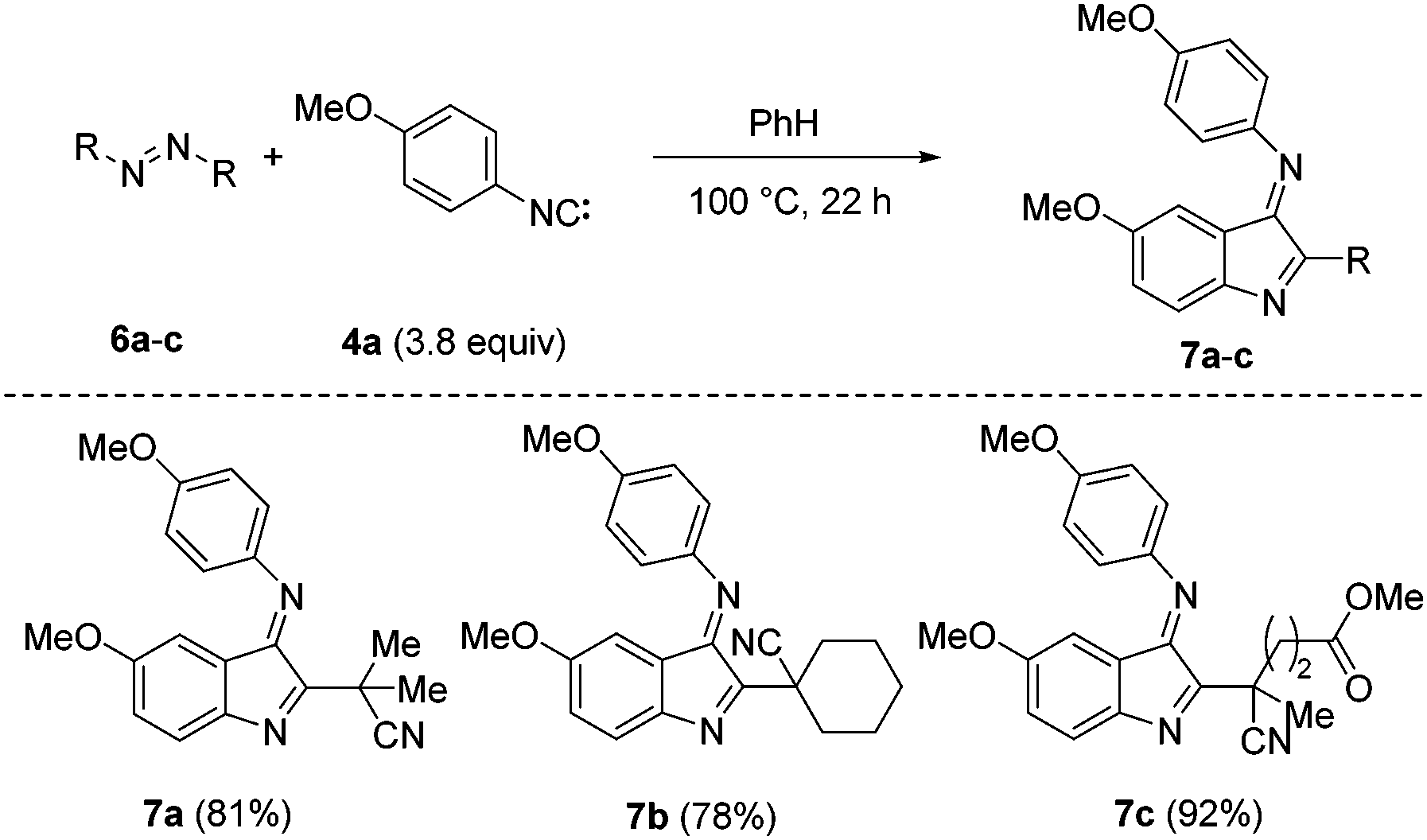 | ||
| Fig. 4 Synthesis of indole imines using azo compounds. | ||
Notably, all of the indole imines 5 and 7 show a very intense colour. For example, the indol-3-imine 5a with methoxy-substituents at the aromatic moiety has a deep red colour (λmax = 478 nm) and the dimethylamino congener 5d shows an intense violet colour (λmax = 564 nm). The UV/vis spectra of these compounds are included in the ESI.† Quantum chemical calculations for these compounds reproduce this difference in absorption very well (λmax (5a) = 479 nm, λmax (5d) = 549 nm with ADC(2)). According to our calculations, these absorption bands are caused by the two lowest-energy singlet–singlet excitations, which are dominated by HOMO → LUMO and (HOMO–1) → LUMO orbital transitions. Their energetic positions are strongly dependent on the electronic nature of the substituents (see the ESI† for details of the calculations).
We next investigated the reactivity of the product indoles using 5a and 7a as substrates. Hydrogenation in ethyl acetate at room temperature (H2, Pd(C)) provided the corresponding 3-arylamino-2-(trifluoromethyl)-1H-indole 8a and 2-methyl-2-(3-(phenylamino)-1H-indol-2-yl)propanenitrile 8b in 80% and 83% yield, respectively (Fig. 5).
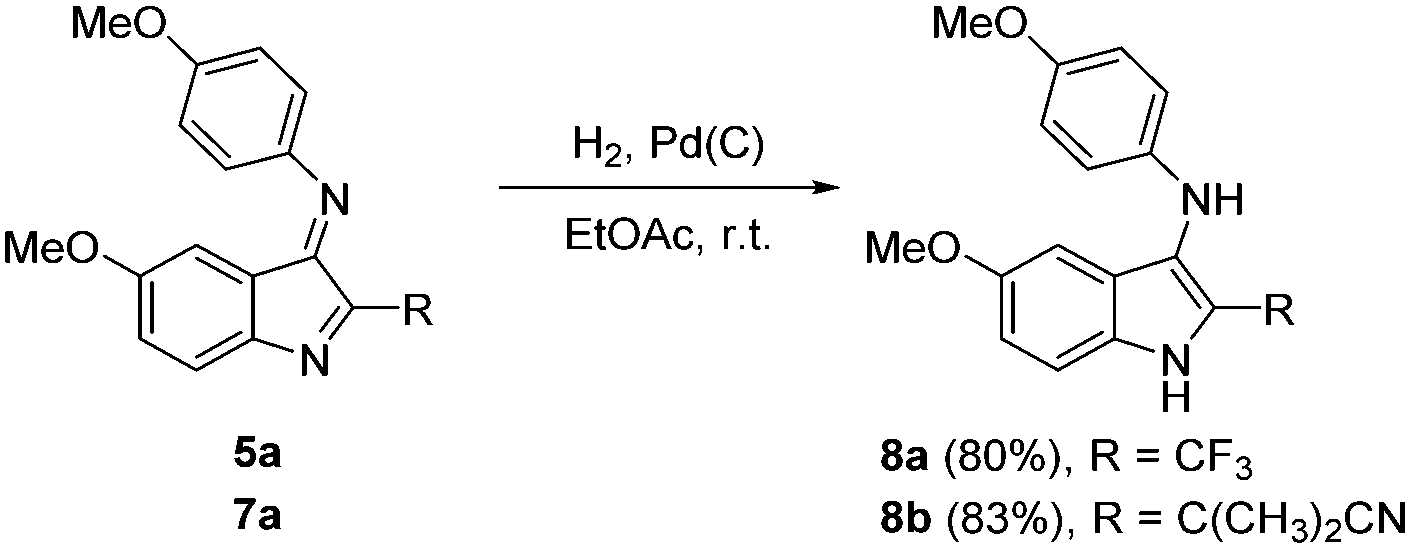 | ||
| Fig. 5 Hydrogenation of 5a and 7a. | ||
Reaction of 5a with para-toluenesulfonic acid monohydrate (1.0 equiv.) in diethyl ether at an elevated temperature (sealed tube) provided quantitatively half aminal 9a, showing that the endocyclic imine functionality is more reactive (Fig. 6). Hydrolysis of the exocyclic imine can be achieved upon treatment with aqueous HCl (see 10a, 99%). Butyllithium undergoes addition with complete regioselectivity to the endocyclic imine and subsequent hydrolysis of the remaining imine with aq. HCl leads to 3-oxindoles, as documented by the successful preparation of 11a, 11d, and 11e.22
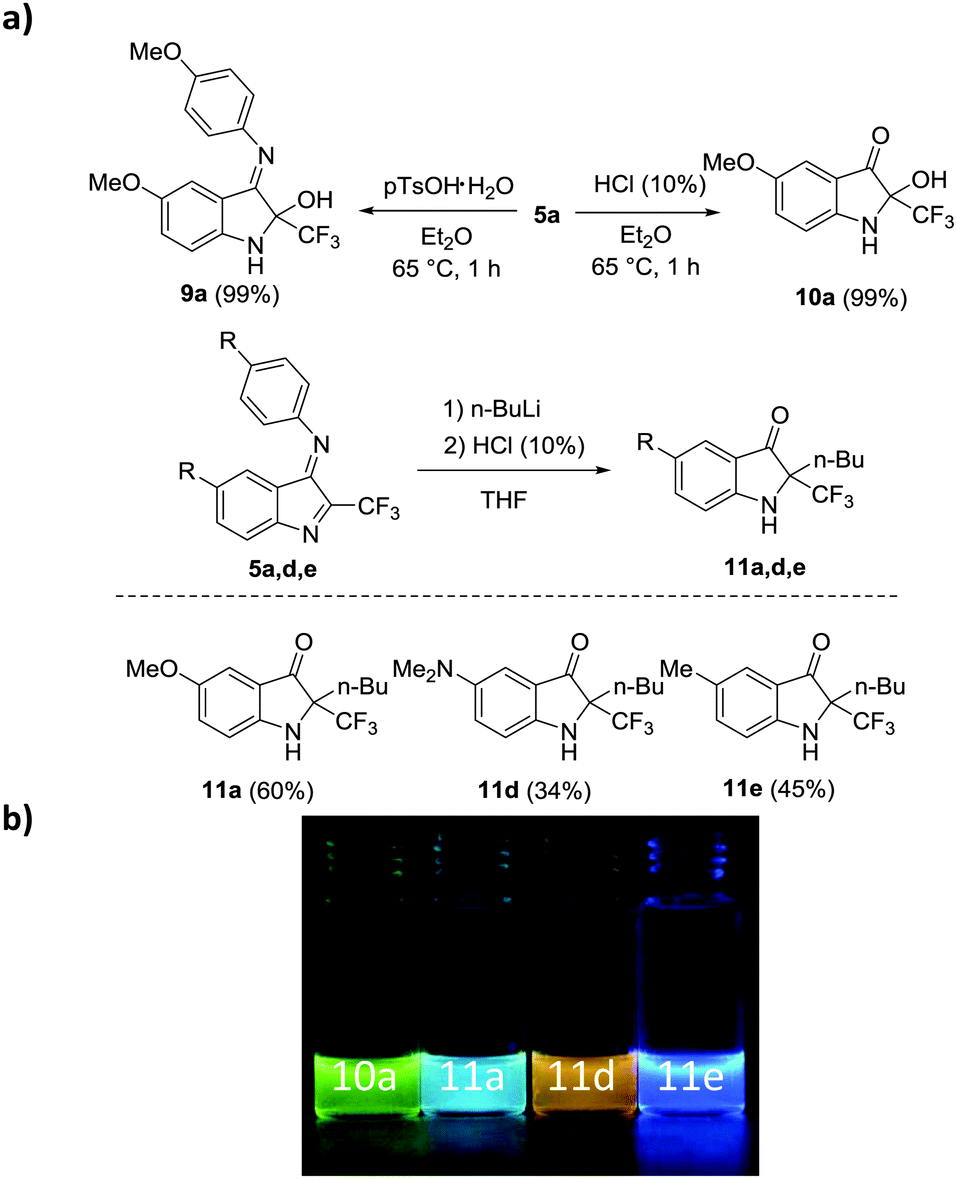 | ||
| Fig. 6 (a) Follow-up chemistry (b) observed fluorescence under UV excitation (365 nm). | ||
Whereas the parent indole imines 5 and 7 show intense colour but no fluorescence, all 3-oxindoles prepared are photoluminescent.23 Notably, imine 9a is not fluorescent indicating that the keto functionality in these compounds is important to attain photoluminescence. For hemiaminal 10a we observed strong green emission and a light blue emission was measured for compound 11a. As expected, substituents at the indole moiety influence the emission properties: the amino derivative 11d shows an orange and the methyl derivative 11e a deep blue emission.
The fluorescent species have been further characterized in terms of absorption spectroscopy and photoluminescence quantum yields, as well as fluorescence excitation, emission, and excited state lifetimes in the solid state, in solution at room temperature and in frozen glassy matrices at 77 K (the results can be found in the ESI†). A summary of the photophysical data is listed in Table 2. The broad, unstructured emission bands reveal a significant charge-transfer character for the excited states, as opposed to vibrationally resolved π–π states. Consequently, strong π-donors generate significant red-shifts. Interestingly, no significant blue-shifts can be traced in frozen matrices at 77 K, even though the solvent molecules cannot rearrange their dipole moments to stabilize the excited states. It should be noted that a higher push–pull character has a detrimental effect on the fluorescence quantum yield, which can be related to a stronger solvent coupling both in the excited as well as in the ground state. In the solid state, the quantum yields drop significantly, which can be ascribed to intermolecular interactions (see Table 2).
| 10a | 11a | 11d | 11e | ||
|---|---|---|---|---|---|
| λ abs [nm] | 420 | 408 | 447 | 392 | |
| (ε [M−1 cm−1]) | (3309) | (3742) | (361) | (3182) | |
| λ em [nm] | Solution | 520 | 488 | 557 | 447 |
| Solid | 574 | 486 | 579 | 511 | |
| 77 K | 501 | 475 | 540 | 450 | |
| τ (±0.05) [ns] | Solution | 14.65 | 15.08 | 14.96 | 8.31 |
| Solid | 1.71 | 1.74 | 4.79 | 2.72 | |
| 0.67 | 0.90 | 0.14 | 9.76 | ||
| 77 K | 21.62 | 17.58 | 24.20 | 12.81 | |
| 33.95 | 6.91 | ||||
| Φ em (±0.02) | Solution | 0.36 | 0.49 | 0.29 | 0.58 |
| Solid | 0.02 | 0.04 | 0.01 | 0.03 |
In summary, we have presented a straightforward access to 2-alkylated indol-3-imines via reaction of (perfluoro)alkyl radicals with two equivalents of an aryl isonitrile. The radical cascade comprises three C–C bond formations. As radical precursors perfluoroalkyl-iodine(III) reagents can be used and LiI is applied as the initiator for these electron-catalyzed processes. Alternatively, AIBN or its derivatives can be applied as C-radical precursors. The product imines show intense colour. The value of the method has been documented by some follow-up reactions. Importantly, hydrolysis of the exocyclic imine leads to 3-oxindoles that show fluorescence properties.
This work was supported by the Deutsche Forschungsgemeinschaft (DFG).
Notes and references
- J.-H. Lee and J. Lee, FEMS Microbiol. Rev., 2010, 34, 426 CrossRef CAS PubMed.
- (a) J. E. Saxton, Nat. Prod. Rep., 1993, 10, 349 RSC; (b) The Chemistry of Heterocyclic Compounds, ed. E. C. Taylor and J. E. Saxton, Wiley-Interscience, New York, 1983/1994, vol. 25 Search PubMed; (c) R. Sundberg, Indoles, Academic Press, New York, 1996 Search PubMed; (d) T. Kawasaki and K. Higuchi, Nat. Prod. Rep., 2005, 22, 761 RSC; (e) N. Saracoglu, Top. Heterocycl. Chem., 2007, 11, 145 Search PubMed; (f) J. J. Li and G. W. Gribble, in Palladium in Heterocyclic Chemistry, ed. J. E. Baldwin and F. R. S. M. R. Williams, Pergamon Press, New York, 2000, vol. 20 Search PubMed.
- Recent reviews regarding the synthesis and modification of indoles, see: (a) S. Cacchi and G. Fabrizi, Chem. Rev., 2005, 105, 2873 CrossRef CAS PubMed; (b) G. R. Humphrey and J. T. Kuethe, Chem. Rev., 2006, 106, 2875 CrossRef CAS PubMed; (c) S. Cacchi and G. Fabrizi, Chem. Rev., 2011, 111, PR215 CrossRef PubMed; (d) N. Yoshikai and Y. Wei, Asian J. Org. Chem., 2013, 2, 466 CrossRef CAS; (e) X.-F. Xiao, H. Neumann and M. Beller, Chem. Rev., 2013, 113, 1 CrossRef PubMed; (f) A. V. Karchava, F. S. Melkonyan and M. A. Yurovskaya, Chem. Heterocycl. Compd., 2012, 48, 391 CrossRef CAS; (g) M. Bandini and A. Eichholzer, Angew. Chem., Int. Ed., 2009, 48, 9608 CrossRef CAS PubMed; (h) G. Bartoli, G. Bencivenni and R. Daipozzo, Chem. Soc. Rev., 2010, 39, 4449 RSC . For selected examples, see: ; (i) S. Kirchberg, R. Fröhlich and A. Studer, Angew. Chem., Int. Ed., 2010, 49, 4235 CrossRef PubMed; (j) V. Ramella, Z. He, C. G. Daniliuc and A. Studer, Org. Lett., 2015, 17, 664 CrossRef CAS PubMed; (k) H. Tokuyama, T. Yamashita, M. T. Reding, Y. Kaburagi and T. Fukuyama, J. Am. Chem. Soc., 1999, 121, 3791 CrossRef CAS; (l) S. Beaumont, V. Pons, P. Retailleau and R. H. Dauban, Angew. Chem., Int. Ed., 2010, 49, 1634 CrossRef CAS PubMed; (m) T. W. Greulich, C. G. Daniliuc and A. Studer, Org. Lett., 2015, 17, 254 CrossRef CAS PubMed.
- (a) K. Müller, C. Feah and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC.
- (a) G. C. B. Harriman, K. G. Carson, D. L. Flynn, M. E. Solomon, Y. Song, B. K. Trivedi, B. D. Roth, C. N. Kolz, L. Pham and K.-L. Sun, Pat., WO2002072549, 2002 Search PubMed; (b) M. T. Baker and M. N. Attala, Pat., WO2003070177, 2003 Search PubMed; (c) M. A. Akanmu, C. Songkram, H. Kagechika and K. Honda, Neurosci. Lett., 2004, 364, 199 CrossRef CAS PubMed; (d) W. H. Romines, R. S. Kania, J. Lou, M. R. Collins, S. R. Cripps, M. He, R. Zhou, C. L. Palmer and J. G. Deal, Pat., WO2003106462, 2003 Search PubMed; (e) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422 CrossRef CAS PubMed.
- Recent examples: (a) V. M. Muzalevskiy, V. G. Nenajdenko, A. V. Shastin, E. S. Balenkova and G. Haufe, Tetrahedron, 2009, 65, 7553 CrossRef CAS; (b) H. Jiang, Y. Wang, W. Wan and J. Hao, Tetrahedron, 2010, 66, 2746 CrossRef CAS; (c) B. I Usachev, D. L. Obydennov and V. Y. J. Sosnovskikh, J. Fluorine Chem., 2012, 135, 278 CrossRef; (d) Z. Chen, J. Zhu, H. Xie, S. Li, Y. Wu and Y. Gong, Adv. Synth. Catal., 2011, 353, 325 CrossRef CAS.
- (a) W. R. Dolbier Jr., Chem. Rev., 1996, 96, 1557 CrossRef; (b) W. R. Dolbier Jr, in Top. Curr. Chem., ed. R. D. Chambers, Springer, Berlin, 1997, vol. 192 Search PubMed; (c) A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8950 CrossRef CAS PubMed.
- B. Zhang, C. Mück-Lichtenfeld, C. G. Daniliuc and A. Studer, Angew. Chem., Int. Ed., 2013, 52, 10792 CrossRef CAS PubMed.
- Y. Cheng, H. Jiang, Y. Zhang and S. Yu, Org. Lett., 2013, 15, 5520 CrossRef CAS PubMed.
- (a) B. Zhang and A. Studer, Chem. Soc. Rev., 2015, 44, 3505 RSC; (b) I. Ryu, N. Sonoda and D. P. Curran, Chem. Rev., 1996, 96, 177 CrossRef CAS PubMed; (c) M. Tobisu and K. Koh, Angew. Chem., Int. Ed., 2012, 51, 11363 CrossRef CAS PubMed; (d) D. Leifert, C. G. Daniliuc and A. Studer, Org. Lett., 2013, 15, 6286 CrossRef CAS PubMed; (e) H. Jiang, Y. Cheng, R. Wang, M. Zheng, Y. Zhang and S. Yu, Angew. Chem., Int. Ed., 2013, 50, 13289 CrossRef PubMed.
- Isonitriles as radical acceptors: (a) T. Saegusa, Y. Ito, S. Kobayashi and K. Hirota, J. Am. Chem. Soc., 1967, 89, 2240 CrossRef CAS; (b) D. Nanni, P. Pareschi, C. Rizzaoli, P. Sgarabotto and A. Tundo, Tetrahedron, 1995, 51, 9045 CrossRef CAS; (c) D. P. Curran, S.-B. Ko and H. Josien, Angew. Chem., Int. Ed. Engl., 1995, 34, 2683 CrossRef CAS; (d) B. Janza and A. Studer, Org. Lett., 2006, 8, 1875 CrossRef CAS PubMed.
- B. Zhang and A. Studer, Org. Lett., 2014, 16, 1216 CrossRef CAS PubMed.
- J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650 CrossRef CAS PubMed.
- (a) M. S. Wiehn, E. V. Vinogradova and A. Togni, J. Fluorine Chem., 2010, 131, 951 CrossRef CAS; (b) R. Shimizu, H. Egami, T. Nagi, J. Chae and Y. Hamashima, Tetrahedron Lett., 2010, 51, 5947 CrossRef CAS; (c) N. Iqbal, S. Choi, E. Ko and E. J. Cho, Tetrahedron Lett., 2012, 53, 2005 CrossRef CAS; (d) E. Mejía and A. Togni, ACS Catal., 2012, 2, 521 CrossRef; (e) Y. Girard, J. G. Atkinson, P. C. Belanger, J. J. Fuentes, J. Rokach and C. S. Rooney, J. Org. Chem., 1983, 48, 3220 CrossRef CAS; (f) D. A. Nagib and D. W. C. MacMillan, Nature, 2011, 480, 224 CrossRef CAS PubMed; (g) M. Yoshida, T. Yoshida, M. Kobayashi and N. Kamigata, J. Chem. Soc., Perkin Trans. 1, 1989, 909 RSC; (h) Q.-Y. Chen and Z.-T. Li, J. Chem. Soc., Perkin Trans. 1, 1993, 645 RSC; (i) T. Kino, Y. Nagase, Y. Ohtsuka, K. Yamamoto, D. Uraguchi, K. Tokuhisa and T. Yamakawa, J. Fluorine Chem., 2010, 131, 98 CrossRef CAS.
- There is only a single example on the radical double isonitrile addition. A tBu-radical, derived from tBuHgCl, reacts with PhNC in a low yield to 2-t-butylindole-3-imine, see: (a) G. A. Russell, R. Rajaratnam and P. Chen, Acta Chem. Scand., 1998, 52, 528 CrossRef CAS . Non radical process: ; (b) Y. Tian, L. Tian, C. Li, X. Jia and J. Li, Org. Lett., 2016, 18, 840 CrossRef CAS PubMed.
- C. Grundmann, Chem. Ber., 1958, 91, 1380 CrossRef CAS.
- (a) A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2011, 50, 5018 CrossRef CAS PubMed; (b) S. Wertz, D. Leifert and A. Studer, Org. Lett., 2013, 15, 928 CrossRef CAS PubMed.
- (a) A. Studer and D. P. Curran, Nat. Chem., 2014, 6, 765 CrossRef CAS PubMed; (b) A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58 CrossRef CAS PubMed.
- D. Leifert and A. Studer, Org. Lett., 2015, 17, 386 CrossRef CAS PubMed.
- Yield is calculated by assuming that one molecule of AIBN leads theoretically to one product molecule, see: A. L. J. Beckwith, V. W. Bowry, W. R. Russell, E. Mann, J. Parr and J. M. D. Storey, Angew. Chem., Int. Ed., 2004, 43, 95 CrossRef PubMed.
- (a) J. Lalevée and J. P. Fouassier, in Encyclopedia of Radicals in Chemistry, Biology and Materials, ed. C. Chatgilialoglu and A. Studer, Wiley, Chichester, UK, 2012, p. 42 Search PubMed The Chemistry of Radical Polymerization, ed. G. Moad and D. H. Solomon, Elsevier, New York, 2006, pp. 60–77 Search PubMed (b) In the corrected yields we are taking into account that in the thermal decomposition of the azo compounds 8 the yield of a free radical is just 60% due to the dimerization in the solvent cage.
- (a) C. Berti and L. Greci, J. Org. Chem., 1981, 46, 3060 CrossRef CAS; (b) Y. Liu and W. W. McWhorter jr., J. Org. Chem., 2003, 68, 2618 CrossRef CAS PubMed; (c) W. Ding, Q.-Q. Zhou, J. Xuan, T.-R. Li, L.-Q. Lu and W.-J. Xiao, Tetrahedron Lett., 2014, 55, 4648 CrossRef CAS.
- (a) Y. Goriya and C. V. Ramana, Chem. Commun., 2013, 49, 6376 RSC; (b) S. Lerch, L.-N. Unkel and M. Brasholz, Angew. Chem., Int. Ed., 2014, 53, 6558 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, and spectroscopic and crystallographic data as well as computational details are included. See DOI: 10.1039/c6cc02284g |
| This journal is © The Royal Society of Chemistry 2016 |