
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Creating a hyperpolarised pseudo singlet state through polarisation transfer from parahydrogen under SABRE†‡
Alexandra M.
Olaru§
a,
Soumya S.
Roy§
a,
Lyrelle S.
Lloyd
a,
Steven
Coombes
b,
Gary G. R.
Green
c and
Simon B.
Duckett
 *a
*a
aDepartment of Chemistry, University of York, Heslington, York YO10 5DD, UK. E-mail: simon.duckett@york.ac.uk; Tel: +44 1904 322564
bAstraZeneca R&D Pharmaceutical Development, Silk Road Business Park, Charter Way, Macclesfield, Cheshire SK10 2NA, UK
cYork Neuroimaging Centre, The Biocentre, York Science Park, Innovation Way, Heslington, York YO10 5DD, UK
First published on 19th May 2016
Abstract
The creation of magnetic states that have long lifetimes has been the subject of intense investigation, in part because of their potential to survive the time taken to travel from the point of injection in a patient to the point where a clinically diagnostic MRI trace is collected. We show here that it is possible to harness the signal amplification by reversible exchange (SABRE) process to create such states in a hyperpolarised form that improves their detectability in seconds without the need for any chemical change by reference to the model substrate 2-aminothiazole. We achieve this by transferring Zeeman derived polarisation that is 1500 times larger than that normally available at 400 MHz with greater than 90% efficiency into the new state, which in this case has a 27 second lifetime.
As magnetic resonance detection, in the forms of NMR and MRI, is relatively insensitive, significant attention is currently placed on employing hyperpolarisation to create observable states that lie far from their normal Boltzmann derived intensity. The resulting signal enhancements offer opportunities to improve the diagnostic power of MRI through the collection of in vivo metabolite time course maps that link spatially resolved measurement to spectroscopic information.1 Such developments require the establishment of protocols to produce hyperpolarised contrast agents with magnetic state lifetimes that facilitate their detection after traveling to the region of clinical interest in the body. Longer magnetic state lifetimes would also be of benefit to NMR spectroscopy more generally as they would allow slower reactions to be followed directly by magnetisation transfer methods.
Whilst hyperpolarised tracers have been produced by brute force methods,2 optical pumping3,4 and dynamic nuclear polarization,5 in this study, parahydrogen (p-H2) derived hyperpolarisation is used as pioneered by Weitekamp,6 Eisenberg7 and Bargon.8,9 The specific process utilised in this paper stems from a 2009 report that showed how magnetization transfer from p-H2 into a substrate could be achieved without the need for any chemical modification of the substrate.10 This process is termed SABRE (signal amplification by reversible exchange).
To date, SABRE has been applied to a range of molecules11–13 including pyridine,14 nicotinamide15 and quinoline,16 and a range of nuclei.17–20 A large number of variables affect the efficiency of SABRE21,22 which include relaxation, temperature and concentration of the catalyst, substrate and p-H2. However, as we are dealing with a hyperpolarisation process rather than a chemical transformation, the strength of the magnetic field experienced during polarisation transfer is also important, as is the number of protons and spin topology of the analytes that are to receive hyperpolarisation.9
Optimisation of the SABRE process is further complicated by the array of magnetic states that are generated, which include longitudinal single-spin (Zeeman polarisation), two-spin and higher order terms.23,24 These states will be characterised by their own unique relaxation times and readout strategies. We show here that it is possible to store this highly polarized Zeeman state in longer lived singlet (LLS) order for subsequent detection. In this context, singlet states are non-magnetic spin isomers of a coupled spin-1/2 pair which are immune to external dipolar coupling and hence often much longer lived (TLLS) than a normal T1 lifetime.25,26 2-Aminothiazole (atz) of Fig. 1 is used in this model study.
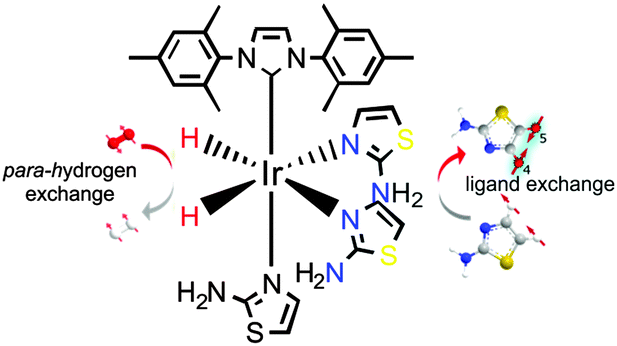 | ||
| Fig. 1 SABRE magnetisation transfer concept based on 3a where ligand exchange leads to hyperpolarised atz. | ||
Its heterocyclic amine core features heavily in biocides, fungicides and dyes, and recently it has been used as a building block in drugs that show wide ranging activity.27–29 This makes atz an attractive candidate for hyperpolarisation because the improved sensitivity may allow it to act as a small molecule probe for metabolic studies.30,31 2-Aminothiazole contains four protons but because these studies are carried out in d4-methanol those of the NH2 group are removed by hydrogen–deuterium exchange. This process leaves behind the two mutually coupled and magnetically isolated H-4 and H-5 protons that are connected by a 3JHH coupling of 3.8 Hz and separated by 160.8 Hz at 400 MHz. Whilst these resonances lie relatively far apart in frequency terms, thereby limiting the lifetime of any shared long-lived state, atz represents a simple test system that builds from the pioneering work of Levitt and is suitable for SABRE.25,26
Since atz possesses two potential metal-ligand binding sites we first set out to characterise the product formed upon reaction with IrCl(COD)(NHC) (1) where the N-heterocyclic carbene (NHC) is derived from 1,3-bis(2,4,6-trimethylphenyl)-imidazolium (1a), 1,3-bis(2,4,6-trimethylphenyl)-4,5-dihydroimidazolium (1b) and 1,3-bis(2,6-diisopropylphenyl)imidazolium (1c) and COD is cyclooctadiene. The formation of [Ir(COD)(NHC)(atz)]Cl (2) proceeds over 5 minutes at room temperature with the binding of atz taking place through the thiazole-ring-nitrogen. When H2 was added to these solutions, the rapid conversion of 2 into [Ir(H)2(NHC)(atz)3]Cl (cis–cis-3) occurs where atz binds similarly. The characterisation of these complexes is described in the ESI.‡
The hyperpolarisation of atz under SABRE was then explored in a 5 mm NMR tube containing 3 bar p-H2 pressure. When a 5 mM solution of 3 was examined in the presence of a 17-fold excess of atz, its H-4 signal exhibited 199 ± 12-, 386 ± 20- and 209 ± 21-fold enhancements for 3a, 3b and 3c respectively after transfer at 60 G. These values changed to 386 ± 82-, 432 ± 38- and 108 ± 12-fold when the atz excess was reduced to 2-fold. These data show that 3b is the most suitable catalyst for magnetisation transfer under SABRE into atz. This observation is supported by the measurement of the build-up rate constant of atz in solution after dissociation from the site trans to hydride of 3a which proved to be 2.04 ± 0.01 s−1 at 300 K which is 15 times slower than the corresponding rate of pyridine build-up in solution from [Ir(IMes)(H)2(py)3]Cl (4), a complex that is know to yield very good pyridine proton signal enhancements under SABRE. The corresponding rate of build-up in 3b was 4.70 ± 0.05 s−1 under similar conditions and suggests that it's greater SABRE efficiency stems from faster ligand exchange.
The corresponding activation parameters for atz build-up in solution and H2 loss in 3 are presented in Table 1. These data suggest that the binding energy of atz in 3a and 3b is lower than that of pyridine in 4 for which the site trans to hydride has a ΔH‡(build-up) value of 95 ± 1 kJ mol−1. Given that the reported pKa of pyridine is 5.17 whilst that of atz is 5.39, it is likely that steric effects act to lengthen the atz bond in the ground state and thereby reducing the subsequent entropy gain of ligand release in order to account for these values.
| Ligand exchange process | H2 loss from | Free atz build-up | ||
|---|---|---|---|---|
| 3a | 3b | 3a | 3b | |
| Raw rate s−1 (300 K) | 0.86 ± 0.01 | 3.41 ± 0.02 | 2.04 ± 0.01 | 4.70 ± 0.05 |
| ΔH‡/kJ mol−1 | 83 ± 3 | 87 ± 2 | 86 ± 3 | 77 ± 3 |
| ΔS‡/J K−1 mol−1 | 37 ± 8 | 62 ± 8 | 54 ± 11 | 31 ± 10 |
| ΔG‡300/kJ mol−1 | 72.2 ± 0.1 | 68.8 ± 0.1 | 70.1 ± 0.1 | 67.9 ± 0.1 |
As we are equilibrating spin-polarisation between hydride and ligand sites during SABRE, we also studied the effect of 3a and 3b on the relaxation times of the states Iz (H-4), Sz (H-5) and the more unusual singlet spin order (TLLS, I·S) of atz as shown in Table 2; these polarisation data were recorded in an automated flow mode as described previously.15 The pulse sequences detailed in the SI were used to make these measurements. The corresponding three relaxation times for free atz are 23, 26 and 27 seconds respectively. Based on the relaxation data of Table 2 it is clear that the presence of 3 acts to redistribute the magnetisation between the coupled spins through the SABRE effect. This change manifests itself in a 65% reduction in the T1 value of H-4 when compared to that of the free material in the presence of 3a; this is much larger than the H-5 site fall of 29%. This change dovetails with the fact that proton H-4 is next to the binding site and experiences the strongest hydride ligand interaction.
| Complex | 3a | 3b | |||
|---|---|---|---|---|---|
3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :atz ratio :atz ratio |
1:2 |
1:17 |
1:2 |
1:17 |
|
| Signal gain | H-4 | −456 | −113 | −441 | −356 |
| H-5 | −693 | −127 | −547 | −425 | |
| T1 (s) | H-4 | 10.0 ± 0.4 | 15.1 ± 0.3 | 12.2 ± 0.5 | 16.2 ± 0.6 |
| H-5 | 18.5 ± 0.6 | 22.9 ± 0.3 | 23.7 ± 0.8 | 25.0 ± 0.8 | |
| TLLS (s) | 16.5 ± 3.2 | 18.3 ± 2.8 | 13.3 ± 2.6 | 26.8 ± 3.9 | |
In order to harness these effects to probe the resulting singlet spin order, a single scan 1H NMR spectrum of atz was collected for comparison purposes that employs thermally equilibrated polarisation at 400 MHz as shown in Fig. 2(a). Fig. 2(b) shows the resulting NMR trace that was produced upon observation of the S0 state after a one second spin-lock and its creation from the thermally polarized state by the method of Levitt.25 This state proved to have a lifetime of 27 seconds in the absence of oxygen and catalyst but was reduced in the presence of 3a and H2 as detailed in Table 2, although with a 20-fold ligand excess and 3b there is little effect. This confirms that catalyst design can be used not only to optimise the level of SABRE, but also to control its effect on the relaxation characteristics of the substrate.32
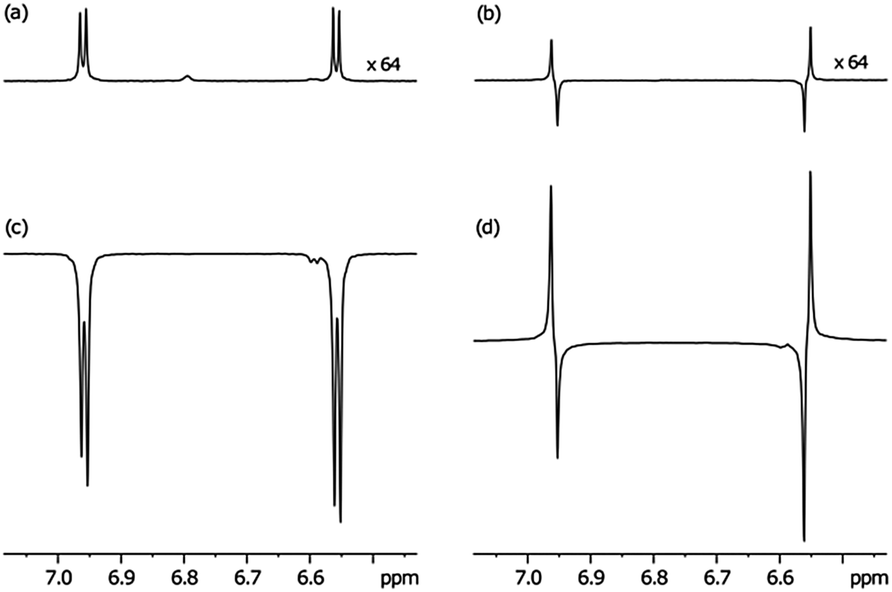 | ||
| Fig. 2 Plots showing the 1H NMR signals of atz at 400 MHz for: (a) a single shot thermally equilibrated 1H NMR spectrum, (b) a S0 derived spectrum of atz that is created and examined 1 s later by the method of Levitt, (c) the corresponding 3a derived SABRE hyperpolarised atz spectrum and (d) the S0 state 1 s after it was created through SABRE and a spin-lock. (Traces in (a) and (b) have a 64 × vertical expansion relative to (c) and (d) respectively). | ||
We then prepared the corresponding hyperpolarised Zeeman spin order in a sealed NMR tube through the action of SABRE via3a as detailed in Fig. 2(c) for subsequent conversion into the S0 state. We used the sealed tube because it produces higher levels of hyperpolarisation than the flow system. The pulse sequence employed a 1 kHz low power spin-lock during state preservation. As revealed in Fig. 2(d) a dramatic increase in the level of S0 spin order results from this process. Based on these data the relayed SABRE transfer occurs with 90% efficiency. Fig. 3 reveals that significant hyperpolarised-S0 derived magnetisation can be seen, even after 60 seconds.
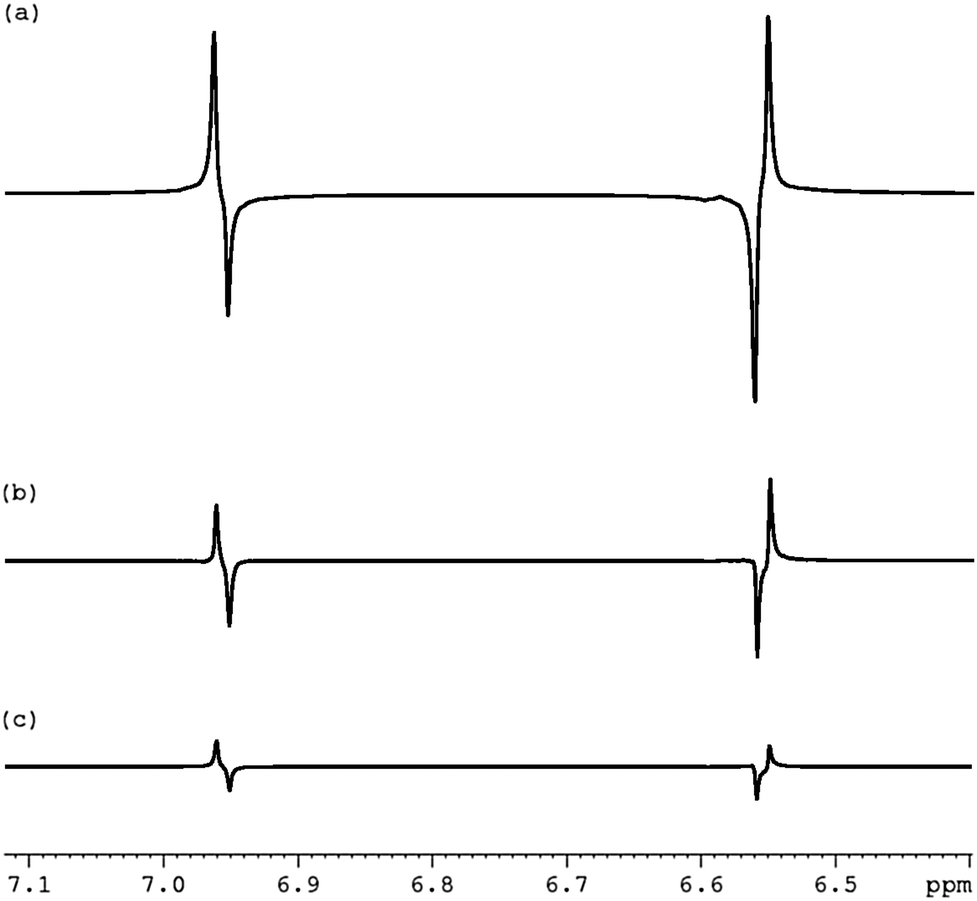 | ||
| Fig. 3 1H NMR traces showing the decay in the SABRE driven singlet (S0) magnetisation over spin-lock times of: (a) 1 s, (b) 30 s and (c) 60 s. | ||
In this report we have demonstrated how SABRE derived hyperpolarisation can be readily used to create long-lived S0 magnetisation by reference to 2-aminothiazole. The efficiency of this process was optimised by varying the SABRE catalyst such that an initial 4.5% level of Zeeman hyperpolarisation (1500-fold enhancement) was harnessed to create the longer-lived S0 magnetisation in 2-aminothiazole. This process reflects a simple and efficient route to rapidly create hyperpolarised long-lived states in a matter of seconds that should be amenable for future in vivo measurement.
We thank the Wellcome Trust (092506 and 098335) and Astra Zeneca (case award LSL) for funding. We are also grateful to discussions with Ryan Mewis, Bruker BioSpin (Joost Lohman, Jean-Max Tyburn, Patrick Krencker and David Kilgour) and Richard. J. Lewis, Michael Bernstein and Christopher J. Sleigh of Astra-Zeneca.
Notes and references
- K. Golman, R. in't Zandt, M. Lerche, R. Pehrson and J. H. Ardenkjaer-Larsen, Cancer Res., 2006, 66, 10855–10860 CrossRef CAS PubMed.
- M. L. Hirsch, N. Kalechofsky, A. Belzer, M. Rosay and J. G. Kempf, J. Am. Chem. Soc., 2015, 137, 8428–8434 CrossRef CAS PubMed.
- A. M. Oros and N. J. Shah, Phys. Med. Biol., 2004, 49, R105–R153 CrossRef CAS PubMed.
- D. M. L. Lilburn, G. E. Pavlovskaya and T. Meersmann, J. Magn. Reson., 2013, 229, 173–186 CrossRef CAS PubMed.
- J. H. Ardenkjær-Larsen, B. Fridlund, A. Gram, G. Hansson, L. Hansson, M. H. Lerche, R. Servin, M. Thaning and K. Golman, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10158–10163 CrossRef PubMed.
- C. R. Bowers and D. P. Weitekamp, J. Am. Chem. Soc., 1987, 109, 5541–5542 CrossRef CAS.
- T. C. Eisenschmid, R. U. Kirss, P. P. Deutsch, S. I. Hommeltoft, R. Eisenberg, J. Bargon, R. G. Lawler and A. L. Balch, J. Am. Chem. Soc., 1987, 109, 8089–8091 CrossRef CAS.
- J. Natterer and J. Bargon, Prog. Nucl. Magn. Reson. Spectrosc., 1997, 31, 293–315 CrossRef.
- R. A. Green, R. W. Adams, S. B. Duckett, R. E. Mewis, D. C. Williamson and G. G. R. Green, Prog. Nucl. Magn. Reson. Spectrosc., 2012, 67, 1–48 CrossRef CAS PubMed.
- R. W. Adams, J. A. Aguilar, K. D. Atkinson, M. J. Cowley, P. I. P. Elliott, S. B. Duckett, G. G. R. Green, I. G. Khazal, J. Lopez-Serrano and D. C. Williamson, Science, 2009, 323, 1708–1711 CrossRef CAS PubMed.
- E. B. Duecker, L. T. Kuhn, K. Muennemann and C. Griesinger, J. Magn. Reson., 2012, 214, 159–165 CrossRef CAS PubMed.
- M. L. Truong, F. Shi, P. He, B. Yuan, K. N. Plunkett, A. M. Coffey, R. V. Shchepin, D. A. Barskiy, K. V. Kovtunov, I. V. Koptyug, K. W. Waddell, B. M. Goodson and E. Y. Chekmenev, J. Phys. Chem. B, 2014, 118, 13882–13889 CrossRef CAS PubMed.
- H. Zeng, J. Xu, J. Gillen, M. T. McMahon, D. Artemov, J.-M. Tyburn, J. A. B. Lohman, R. E. Mewis, K. D. Atkinson, G. G. R. Green, S. B. Duckett and P. C. M. van Zijl, J. Magn. Reson., 2013, 237, 73–78 CrossRef CAS PubMed.
- K. D. Atkinson, M. J. Cowley, P. I. P. Elliott, S. B. Duckett, G. G. R. Green, J. Lopez-Serrano and A. C. Whitwood, J. Am. Chem. Soc., 2009, 131, 13362–13368 CrossRef CAS PubMed.
- R. E. Mewis, K. D. Atkinson, M. J. Cowley, S. B. Duckett, G. G. R. Green, R. A. Green, L. A. R. Highton, D. Kilgour, L. S. Lloyd, J. A. B. Lohman and D. C. Williamson, Magn. Reson. Chem., 2014, 52, 358–369 CrossRef CAS PubMed.
- L. S. Lloyd, R. W. Adams, M. Bernstein, S. Coombes, S. B. Duckett, G. G. R. Green, R. J. Lewis, R. E. Mewis and C. J. Sleigh, J. Am. Chem. Soc., 2012, 134, 12904–12907 CrossRef CAS PubMed.
- V. V. Zhivonitko, I. V. Skovpin and I. V. Koptyug, Chem. Commun., 2015, 51, 2506–2509 RSC.
- M. J. Burns, P. J. Rayner, G. G. R. Green, L. A. R. Highton, R. E. Mewis and S. B. Duckett, J. Phys. Chem. B, 2015, 119, 5020–5027 CrossRef CAS PubMed.
- M. L. Truong, T. Theis, A. M. Coffey, R. V. Shchepin, K. W. Waddell, F. Shi, B. M. Goodson, W. S. Warren and E. Y. Chekmenev, J. Phys. Chem. C, 2015, 119, 8786–8797 CAS.
- R. E. Mewis, R. A. Green, M. C. R. Cockett, M. J. Cowley, S. B. Duckett, G. G. R. Green, R. O. John, P. J. Rayner and D. C. Williamson, J. Phys. Chem. B, 2015, 119, 1416–1424 CrossRef CAS PubMed.
- B. J. A. van Weerdenburg, S. Gloeggler, N. Eshuis, A. H. J. Engwerda, J. M. M. Smits, R. de Gelder, S. Appelt, S. S. Wymenga, M. Tessari, M. C. Feiters, B. Blümich and F. P. J. T. Rutjes, Chem. Commun., 2013, 49, 7388–7390 RSC.
- L. S. Lloyd, A. Asghar, M. J. Burns, A. Charlton, S. Coombes, M. J. Cowley, G. J. Dear, S. B. Duckett, G. R. Genov, G. G. R. Green, L. A. R. Highton, A. J. J. Hooper, M. Khan, I. G. Khazal, R. J. Lewis, R. E. Mewis, A. D. Roberts and A. J. Ruddlesden, Catal. Sci. Technol., 2014, 4, 3544–3554 CAS.
- R. W. Adams, S. B. Duckett, R. A. Green, D. C. Williamson and G. G. R. Green, J. Chem. Phys., 2009, 131, 194505 CrossRef PubMed.
- A. N. Pravdivtsev, A. V. Yurkovskaya, H.-M. Vieth and K. L. Ivanov, Phys. Chem. Chem. Phys., 2014, 16, 24672–24675 RSC.
- M. Carravetta and M. H. Levitt, J. Am. Chem. Soc., 2004, 126, 6228–6229 CrossRef CAS PubMed.
- M. H. Levitt, in Annu. Rev. Phys. Chem., ed. M. A. Johnson and T. J. Martinez, Annual Reviews, Palo Alto, 2012, vol. 63, pp. 89–105 Search PubMed.
- A. S. Kalgutkar, I. Gardner, R. S. Obach, C. L. Shaffer, E. Callegari, K. R. Henne, A. E. Mutlib, D. K. Dalvie, J. S. Lee, Y. Nakai, J. P. O'Donnell, J. Boer and S. P. Harriman, Curr. Drug Metab., 2005, 6, 161–225 CrossRef CAS PubMed.
- I. Lagoja, C. Pannecouque, G. Griffioen, S. Wera, V. M. Rojasdelaparra and A. Van Aerschot, Eur. J. Pharm. Sci., 2011, 43, 386–392 CrossRef CAS PubMed.
- M. Pieroni, B. J. Wan, S. H. Cho, S. G. Franzblau and G. Costantino, Eur. J. Med. Chem., 2014, 72, 26–34 CrossRef CAS PubMed.
- J. Kurhanewicz, D. B. Vigneron, K. Brindle, E. Y. Chekmenev, A. Comment, C. H. Cunningham, R. J. DeBerardinis, G. G. Green, M. O. Leach, S. S. Rajan, R. R. Rizi, B. D. Ross, W. S. Warren and C. R. Malloy, Neoplasia, 2011, 13, 81–97 CrossRef CAS PubMed.
- T. L. Davis, K. K. Kwong, R. M. Weisskoff and B. R. Rosen, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 1834–1839 CrossRef CAS.
- G. Stevanato, J. T. Hill-Cousins, P. Hakansson, S. S. Roy, L. J. Brown, R. C. D. Brown, G. Pileio and M. H. Levitt, Angew. Chem., Int. Ed., 2015, 54, 3740–3743 CrossRef CAS PubMed.
Footnotes |
| † Data created during this research are available by request from the University of York Data Catalogue http://dx.doi.org/10.15124/f0779356-a6f1-4c36-bac4-6fdf280ff6b2. |
| ‡ Electronic supplementary information (ESI) available: NMR measurements and characterisation. See DOI: 10.1039/c6cc02020h |
| § These authors contributed equally to the work of this manuscript. |
| This journal is © The Royal Society of Chemistry 2016 |