
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePropentdyopent: the scaffold of a heme metabolite as an electron reservoir in transition metal complexes†
R.
Gautam
,
T. M.
Chang
,
A. V.
Astashkin
,
K. M.
Lincoln
and
E.
Tomat
*
University of Arizona, Department of Chemistry and Biochemistry, 1306 E. University Blvd., Tucson AZ 85721, USA. E-mail: tomat@email.arizona.edu
First published on 25th April 2016
Abstract
The dipyrrin-1,9-dione scaffold of heme metabolite propendyopent coordinates late transition metals (Co, Ni, Cu, and Zn) forming homoleptic, pseudo-tetrahedral complexes. Electrochemical and spectroscopic studies reveal that the monoanionic, bidentate ligands behave as electron reservoirs as the complexes reversibly host one or two ligand-based radicals.
The degradation of heme in mammalian metabolism begins with the formation of linear tetrapyrrole biliverdin (Chart 1), which is catalysed by heme oxygenase primarily during the processing of senescent erythrocytes. Subsequent enzymatic and non-enzymatic reactions lead to the formation of lower-order oligopyrroles,1,2 containing three or two pyrrole rings, which were first isolated from urinary excretions. The coordination and redox chemistry of these urochrome pigments is far less established than that of their tetrapyrrolic congeners. We recently reported the first transition metal complexes of the tripyrrin-1,14-dione fragment (Chart 1).3 This tridentate planar scaffold serves as a robust platform for Pd(II) coordination and ligand-based one-electron processes, thus behaving as a redox-active ligand for the storage of redox equivalents. In general, heme metabolites could offer both the stability of physiological end products and the rich redox chemistry of non-innocent ligands for the engineering of novel redox reactivity. Herein, we examine the metal coordination and redox properties of the propentdyopent system, a heme metabolite and dipyrrolic analog of tripyrrindione and biliverdin.
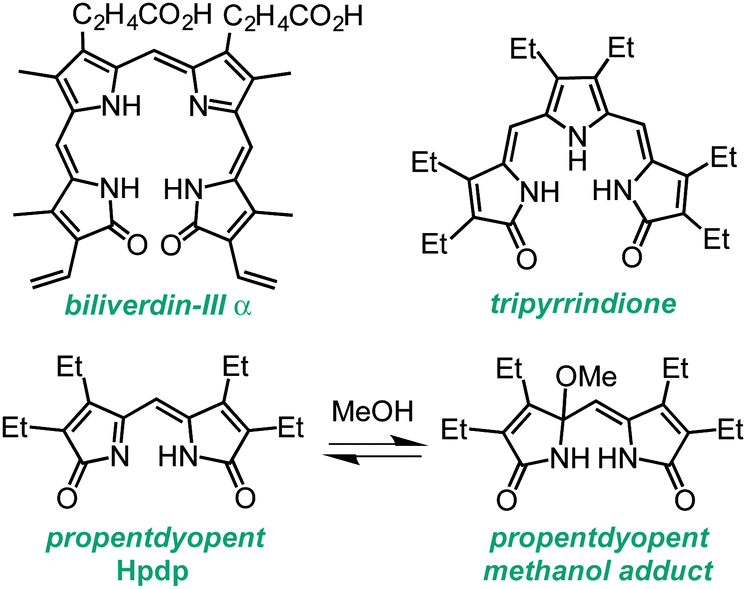 | ||
| Chart 1 Biliverdin pigment derived from heme b and synthetic analogs of smaller fragments of heme degradation. | ||
The unorthodox name propentdyopent refers to the absorption maximum (525 nm) of the red products (pentdyopents) obtained by reduction of these urinary pigments in alkaline solutions, which was first observed by Stokvis in 1870.4 Subsequent studies over a period of five decades established the molecular structure of the propentdyopent family as well as the formation of colorless adducts in the presence of nucleophilic solvents such as water and methanol (Chart 1).5,6
Propentdyopent pigments are structurally related to the bidentate ligands of the large family of dipyrrins,7 which form dipyrrinato metal complexes employed widely in multiple applications including fluorescent labels for bioimaging (i.e., BODIPY dyes),8 coordination polymers,9–11 and catalysts.12 In spite of this similarity with a versatile family of ligands, the coordination chemistry of propentdyopents remains rather unexplored. Early reports indicated the formation of a zinc complex that was not characterized fully.5,13 Interestingly, Cu(II) and Co(II) propentdyopent complexes were isolated upon oxidative degradation of copper14 and cobalt15 complexes of octaethylbilindione, a biliverdin analog. Although propentdyopent complexes have not been detected (and possibly searched for) in biological settings, their formation could therefore arise, at least in principle, from direct metal binding or also from decomposition of complexes of larger pigments. These considerations, along with the possibility to access a new redox-active framework analogous to the tripyrrindione system, motivated the present study.
An all-ethyl analog of the naturally occurring propentdyopents (which typically feature methyl, vinyl or propionate substituents) was prepared from the photo-oxygenation of tetraethyldipyrrinone (aka tetraethylpyrromethenone) and isolated as the methanol adduct Hpdp·MeOH as previously reported.6,16,17 Propentdyopent complexes were prepared in good yields by briefly refluxing (10–30 min) methanolic solutions of Hpdp·MeOH in the presence of chloride or acetate salts of Co(II), Ni(II), Cu(II), and Zn(II) (see ESI† for experimental details). The progress of metal coordination was monitored by UV-visible absorption spectroscopy owing to the π–π* transitions in the visible region that are characteristic of dipyrrin complexes (Fig. S3, ESI†).
Single crystals were obtained for all four complexes and analysed by X-ray diffractometry (Fig. 1). Each neutral, homoleptic complex features two bidentate propentdyopent ligands in a non-planar geometry typical of 1,9-substituted dipyrrins.7,18,19 All complexes are pseudo-tetrahedral, with dihedral angles between the propentdyopent planes being closer to orthogonality for the zinc complex (79.8°) and most distorted for the copper complex (54.2°). These data therefore showed that homoleptic cobalt and copper propentdyopent complexes, which were crystallized previously following degradation of tetrapyrrolic octaethylbilindione complexes,14,15 can be obtained directly from reaction with Hpdp·MeOH. For consistency of comparison across this series of structures of similar resolution, Fig. 1 shows our structures of Co(pdp)2 and Cu(pdp)2 rather than those reported previously (see ESI† for details).
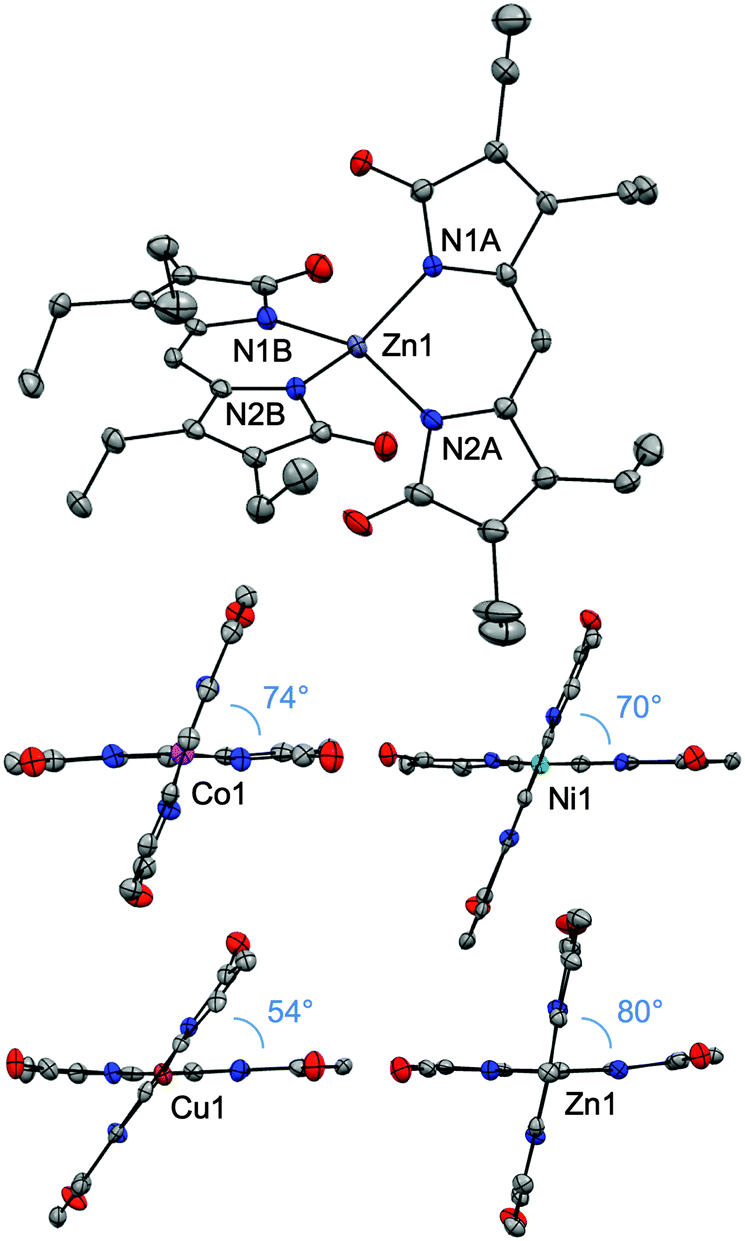 | ||
| Fig. 1 Crystal structures of homoleptic complexes Co(pdp)2, Ni(pdp)2, Cu(pdp)2, and Zn(pdp)2 (CCDC: Co, 1434495; Ni, 1434496; Cu, 1431584; Zn, 1431605). In the views highlighting the dihedral angles between the planes of the ligands, the ethyl substituents have been omitted. Thermal ellipsoids are scaled to the 50% probability level. | ||
Bond lengths on the ligand frameworks (Table S3, ESI†) remain comparable across this series (for instance, with C–O distances in the (1.209(7)–1.232(6) Å) range characteristic of carbonyl double bonds) indicating consistency of electronic structure and redox state for the propentdyopent scaffold, which coordinates as a monoanionic bidentate ligand in all cases. In the isolated neutral complexes, all the metal cations therefore maintain their divalent oxidation states and, unlike the tripyrrindione system,3 Hpdp does not undergo oxidation upon metal coordination. These considerations were confirmed by spectroscopic methods (vide infra).
The 1H NMR spectrum of diamagnetic complex Zn(pdp)2 exhibits a single set of resonances for the pdp− ligands (Fig. S1, ESI†), thus indicating that D2 symmetry is maintained in solution at room temperature. Similarly, only five ligand resonances are observed in the 1H NMR spectrum (Fig. S2, ESI†) of Ni(pdp)2, a pseudo-tetrahedral d8 species (S = 1). As observed for other paramagnetic Ni(II) complexes,20 this spectrum presents fairly sharp peaks over a relatively narrow spectral window (70 ppm), and the resonances for the meso-type protons are strongly shifted upfield (by 59 ppm) relative to those of the corresponding protons in Zn(pdp)2.
The continuous-wave (CW) EPR spectrum of Co(pdp)2 (Fig. S7, ESI†) recorded at 10 K presents a broad signal with turning points at (g⊥, g∥) ≈ (4.5, 2.01) that are characteristic of a Co(II) center in tetrahedral crystal field (S = 3/2). The EPR data also confirmed a divalent oxidation state for the copper center in Cu(pdp)2 (Fig. S8 and S9, ESI†). For both complexes, the lack of hyperfine structure from the central ion nucleus (I = 3/2 and 7/2 for 63,65Cu and 59Co, respectively) in the spectra of samples in frozen glassy solutions indicates structural disorder likely related to a variation of the N–metal–N bond angle for each of the pdp− ligands and the dihedral angle between the ligand planes. In liquid solutions at room temperature, these structural variations are dynamic and rapid within the EPR timescale, which results in exchange narrowing and a resolved hyperfine structure for the Cu(II) complex (Fig. S9, ESI†). The EPR signal from the Co(II) complex is not observed at room temperature because of very short magnetic relaxation times.
All structural and spectroscopic data confirmed a divalent oxidation state for the metal centers in the isolated propentdyopent complexes. Their redox behaviour was then investigated by electrochemical methods (Fig. 2). The cathodic sweep in cyclic voltammograms collected in CH2Cl2 presents two quasi-reversible one-electron events at half-wave potentials lower than −1.0 V vs. Fc/Fc+ (Table S5, ESI†). These processes, which were previously documented (but not assigned) for cobalt propentdyopent complexes,15 are consistent across this series of complexes including the redox-inactive d10 Zn(II) cation and are therefore attributed to ligand-based reduction events. The small difference in half-potentials (180–260 mV) is ascribed to solvation energy effects for the two redox pairs (neutral/monoanionic vs. monoanionic/dianionic species) rather than to delocalization of the first reducing equivalent on both propentdyopent ligands.21 As previously noted for the sequential reduction of homoleptic zinc complexes of pyridine-2,6-diimine22 and formazanate23 ligands, the cathodic waves are thus assigned to localized ligand-based reductions forming [Zn(pdp˙)(pdp)]− and then [Zn(pdp˙)2]2− in the case of the zinc species. In addition, the voltammograms of the complexes of redox-active cations Cu(II) and Ni(II) exhibit one-electron, quasi-reversible oxidative processes, presumably of metal-based character, at 0.93 and 0.74 V, respectively. The Co(II) species presents an irreversible oxidative event at similarly positive potential. Collectively, these electrochemical observations support the notion of bidentate propendyopent ligands serving as electron reservoirs and prompt further characterization of the reduced species. For spectroelectrochemical and EPR spectroscopic investigations, the zinc complex was selected because of its sharp UV-visible absorption bands and because of its lack of metal-based electronic spins.
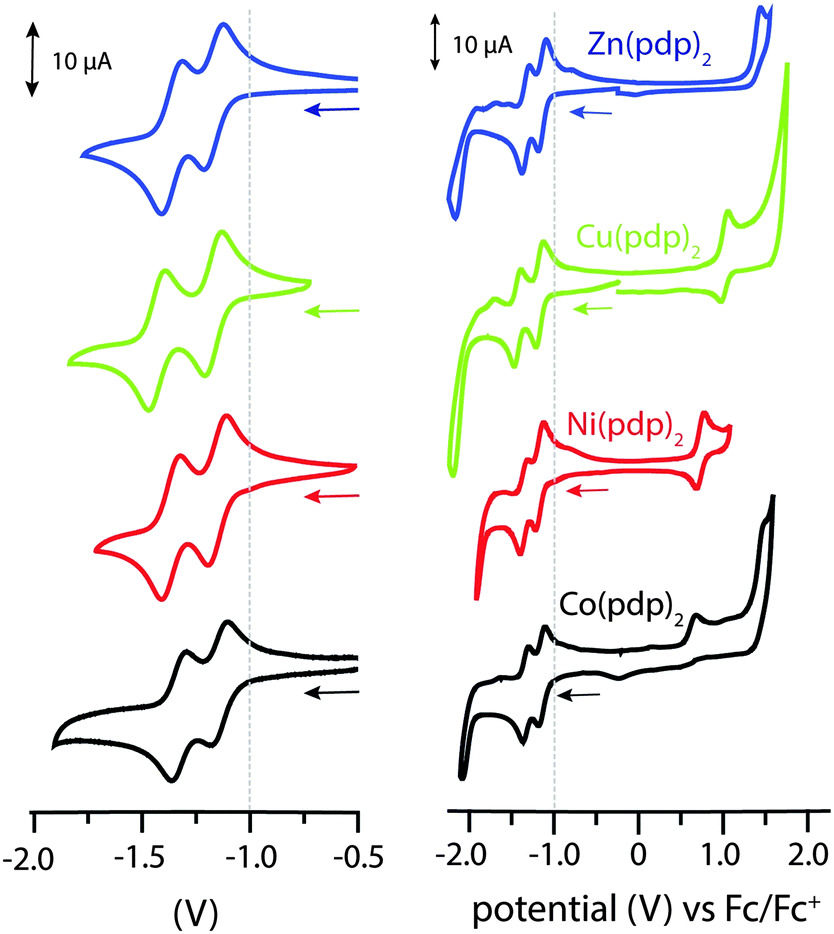 | ||
| Fig. 2 Cyclic voltammograms of Co(pdp)2, Ni(pdp)2, Cu(pdp)2, and Zn(pdp)2 at a glassy carbon electrode in CH2Cl2 with (n-Bu4N)(PF6) as a supporting electrolyte. Data collected at a 100 mV s−1 scan rate using a Ag/AgCl quasi-reference electrode and a platinum wire auxiliary electrode. | ||
As determined by spectroelectrochemical methods, the one-electron reduction of Zn(pdp)2 is accompanied by a decrease of the main absorption bands (361 and 491 nm) and by the appearance of a new π–π* transition at 427 nm (Fig. 3, top panel), which doubles in intensity upon formation of the two-electron reduction product (Fig. 3, bottom panel). The observed additivity of this band (and of the near-IR bands, vide infra) is consistent with two separate one-electron processes occurring on each ligand of the homoleptic complex,22,24 and sequential formation of [Zn(pdp˙)(pdp)]− and [Zn(pdp˙)2]2− with little or negligible interligand delocalization. In addition, the first reduction causes the appearance of two near-IR bands (670 and 740 nm), which are increased in intensity and slightly red-shifted (679 and 748 nm) by the second reduction. These low-energy bands are typical of ligand-based π radicals in oligopyrroles including tripyrrindione,3 corrole,25 2,2′-bidipyrrin26 and bis(phenolate)-dipyrrin.27 Confirming the reversible storage of reducing equivalents, exposure to air (over a period of 4 min) or application of an oxidizing potential yield over 95% of the parent complex (Fig. S6, ESI†).
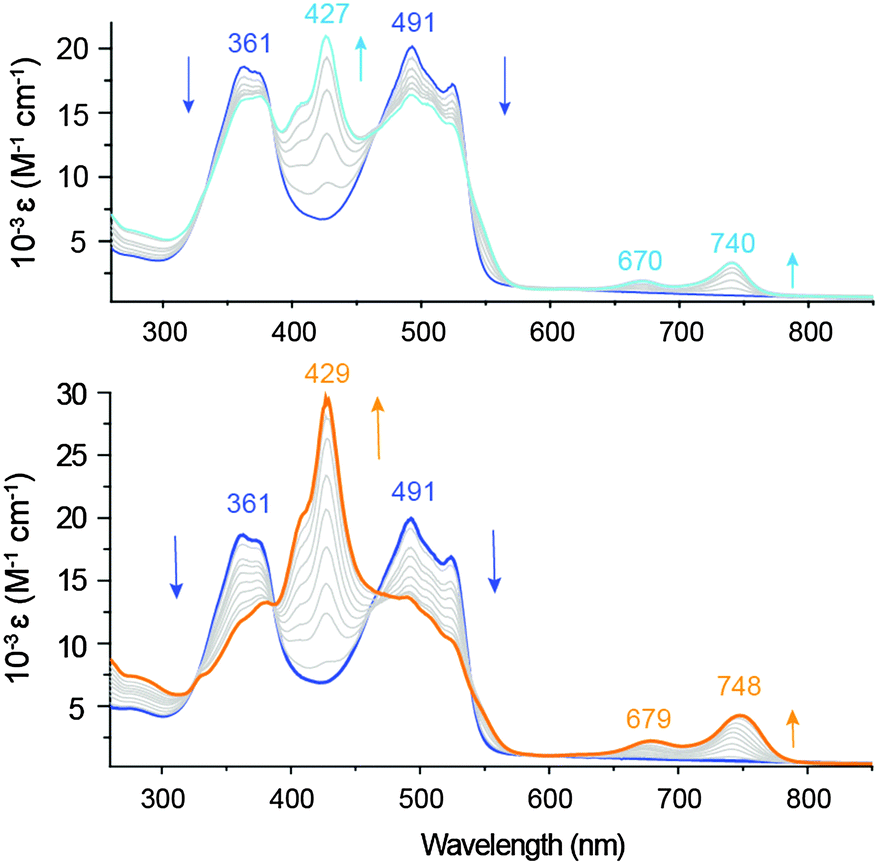 | ||
| Fig. 3 UV-visible absorption spectral changes observed upon reduction of Zn(pdp)2 (67 μM, DMF, 0.1 M [NBu4][PF6]) by controlled potential electrolysis at −1.2 V (200 s) to give [Zn(pdp˙)(pdp)]− (top panel) and at −1.8 V (400 s) to give [Zn(pdp˙)2]2− (bottom panel). | ||
Both the one-electron and the two-electron reduction products of Zn(pdp)2 could be accessed by chemical reduction and detected by EPR spectroscopy. Following addition of a sub-equimolar amount of sodium amalgam (0.8 equiv. Na, 4 h) to a solution of Zn(pdp)2 in THF at room temperature, the EPR spectrum (trace a, Fig. 4) of the reaction mixture presents a narrow line (0.76 mT) at Bo = 336.9 mT (g = 2.003) as expected for [Zn(pdp˙)(pdp)]−, a one-electron reduction product featuring a delocalized ligand-based organic radical (S = 1/2). In the presence of excess reductant (3.8 equiv. Na), the spectrum of a triplet species (S = 1) is clearly seen under a larger magnification (trace b, 2.1 h, Fig. 4). The signal increases in amplitude as the reaction progresses and presumably more reductant becomes available in the heterogeneous mixture (trace c, 3.3 h, Fig. 4).‡ This spectrum results from a diradical species with a splitting of 12.5 mT between the ∥ features. The observed dipole interaction is consistent with diradical species [Zn(pdp˙)2]2− featuring two unpaired electrons, each delocalized on one of the reduced propentdyopent ligands.
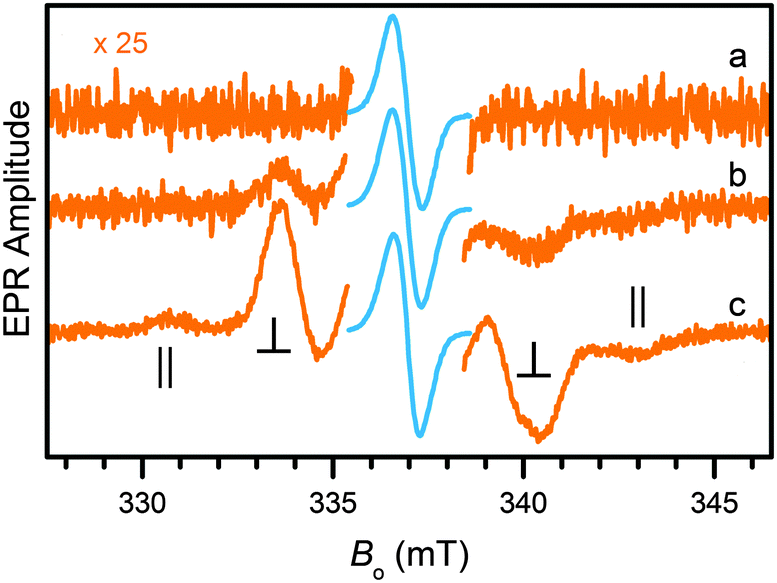 | ||
| Fig. 4 EPR spectra obtained upon reduction of Zn(pdp)2 (0.4–0.6 mM, THF) with Na(Hg) and showing formation of an organic radical in sub-equimolar conditions (trace a, 0.8 equiv. Na, 4 h) and appearance of increasing amounts of a diradical species in the presence of excess reductant (traces b and c, 3.8 equiv. Na, 2.1 and 3.3 h, respectively). The spectra were normalized to the same amplitude of the central line to allow for comparison of the diradical signals (shown in orange). Experimental conditions: microwave frequency, 9.448 GHz; microwave power, 2 mW; field modulation amplitude, 0.6 mT; temperature, 77 K. | ||
In summary, our data show that the propentdyopent scaffold of urinary pigments and heme metabolites serves as a bidentate monoanionic ligand in pseudo-tetrahedral Co(II), Ni(II), Cu(II), and Zn(II) complexes. Notably, electrochemical and spectroscopic studies revealed two quasi-reversible reduction processes of ligand-based character leading to formation of paramagnetic anions [Zn(pdp˙)(pdp)]− and [Zn(pdp˙)2]2− in the case of Zn(pdp)2. A rich ligand-based redox chemistry is not uncommon in metal complexes of oligopyrroles, which typically present frontier orbitals of predominantly ligand character. The electron-rich π system of dipyrrinato ligands, however, is often implicated in one-electron oxidative processes.19,28,29 In contrast, in homoleptic propentdyopent complexes, we demonstrated that electron-rich dipyrrolic platforms can reversibly undergo reduction and thereby serve as electron reservoirs for the storage of two reducing equivalents. These biologically occurring dipyrroles therefore extend the chemistry of dipyrrins and offer ligand-based reactivity of potential relevance to their chemistry in biological settings and to synthetic and catalytic applications.
We gratefully acknowledge Drs Jonathan Loughrey and Sue Roberts for assistance with initial electrochemical measurements and analysis of X-ray diffraction data, respectively. This work was supported by the University of Arizona and by the National Science Foundation (CAREER grant 1454047 to E. T.).
Notes and references
- H. Falk, The Chemistry of Linear Oligopyrroles and Bile Pigments, Springer-Verlag, Wien, 1989 Search PubMed.
- M. Broring, in Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific, Singapore, 2010, vol. 8, ch. 41, p. 343 Search PubMed.
- R. Gautam, J. J. Loughrey, A. V. Astashkin, J. Shearer and E. Tomat, Angew. Chem., Int. Ed., 2015, 14894 CrossRef CAS PubMed.
- H. Fischer and H. F. von Dobeneck, Hoppe-Seyler's Z. Physiol. Chem., 1940, 263, 125 CrossRef CAS.
- R. Bonnett, M. J. Dimsdale and G. F. Stephenson, J. Chem. Soc., Perkin Trans. 1, 1987, 439 RSC.
- R. Bonnett and S. Ioannou, J. Chem. Soc., Chem. Commun., 1986, 213 RSC.
- T. E. Wood and A. Thompson, Chem. Rev., 2007, 107, 1831 CrossRef CAS PubMed.
- T. Kowada, H. Maeda and K. Kikuchi, Chem. Soc. Rev., 2015, 44, 4953 RSC.
- R. Sakamoto, K. Hoshiko, Q. Liu, T. Yagi, T. Nagayama, S. Kusaka, M. Tsuchiya, Y. Kitagawa, W.-Y. Wong and H. Nishihara, Nat. Commun., 2015, 6, 6713 CrossRef CAS PubMed.
- R. Matsuoka, R. Toyoda, R. Sakamoto, M. Tsuchiya, K. Hoshiko, T. Nagayama, Y. Nonoguchi, K. Sugimoto, E. Nishibori, T. Kawai and H. Nishihara, Chem. Sci., 2015, 6, 2853 RSC.
- J. R. Stork, V. S. Thoi and S. M. Cohen, Inorg. Chem., 2007, 46, 11213 CrossRef CAS PubMed.
- E. T. Hennessy, R. Y. Liu, D. A. Iovan, R. A. Duncan and T. A. Betley, Chem. Sci., 2014, 5, 1526 RSC.
- H. von Dobeneck, Hoppe-Seyler's Z. Physiol. Chem., 1941, 269, 268 CrossRef CAS.
- A. L. Balch, M. Mazzanti, B. C. Noll and M. M. Olmstead, J. Am. Chem. Soc., 1993, 115, 12206 CrossRef CAS.
- R. Koerner, M. M. Olmstead, P. M. Van Calcar, K. Winkler and A. L. Balch, Inorg. Chem., 1998, 37, 982 CrossRef CAS.
- R. Bonnett, D. G. Buckley and D. Hamzetash, J. Chem. Soc., Perkin Trans. 1, 1981, 322 RSC.
- R. Bonnett, S. Ioannou and F. J. Swanson, J. Chem. Soc., Perkin Trans. 1, 1989, 711 RSC.
- Q. Miao, J.-Y. Shin, B. O. Patrick and D. Dolphin, Chem. Commun., 2009, 2541 RSC.
- R. Toyoda, M. Tsuchiya, R. Sakamoto, R. Matsuoka, K.-H. Wu, Y. Hattori and H. Nishihara, Dalton Trans., 2015, 44, 15103 RSC.
- A. Sanchez-Mendez, J. M. Benito, E. de Jesus, F. J. de la Mata, J. C. Flores, R. Gomez and P. Gomez-Sal, Dalton Trans., 2006, 5379 RSC.
- P. Zanello, C. Nervi and F. Fabrizi de Biani, Inorganic Electrochemistry: Theory, Practice and Application, RSC, Cambridge, 2011 Search PubMed.
- B. de Bruin, E. Bill, E. Bothe, T. Weyhermuller and K. Wieghardt, Inorg. Chem., 2000, 39, 2936 CrossRef CAS PubMed.
- M.-C. Chang, T. Dann, D. P. Day, M. Lutz, G. G. Wildgoose and E. Otten, Angew. Chem., Int. Ed., 2014, 53, 4118 CrossRef CAS PubMed.
- M. Orio, C. Philouze, O. Jarjayes, F. Neese and F. Thomas, Inorg. Chem., 2010, 49, 646 CrossRef CAS PubMed.
- P. Schweyen, K. Brandhorst, R. Wicht, B. Wolfram and M. Broring, Angew. Chem., Int. Ed., 2015, 54, 8213 CrossRef CAS PubMed.
- M. Broring, C. D. Brandt, J. Bley-Escrich and J. P. Gisselbrecht, Eur. J. Inorg. Chem., 2002, 910 CrossRef CAS.
- A. Kochem, L. Chiang, B. Baptiste, C. Philouze, N. Leconte, O. Jarjayes, T. Storr and F. Thomas, Chem. – Eur. J., 2012, 18, 14590 CrossRef CAS PubMed.
- E. R. King and T. A. Betley, J. Am. Chem. Soc., 2009, 131, 14374 CrossRef CAS PubMed.
- K. Servaty, E. Cauet, F. Thomas, J. Lambermont, P. Gerbaux, J. De Winter, M. Ovaere, L. Volker, N. Vaeck, L. Van Meervelt, W. Dehaen, C. Moucheron and A. Kirsch-De Mesmaeker, Dalton Trans., 2013, 42, 14188 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures and experimental methods for X-ray diffraction, UV-visible absorption, electrochemistry, and EPR spectroscopic analyses. CCDC 1431584, 1431605, 1434495 and 1434496. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc01961g |
| ‡ At longer reaction times, both the central line and the dipolar features gradually change, indicating degradation through ligand dissociation and/or reactivity of the reduction products. These effects occur faster with larger amounts of reductant; however, the initial products are stable for about 4 h at room temperature in the presence of 3.8 equiv. of sodium under an inert atmosphere. |
| This journal is © The Royal Society of Chemistry 2016 |