
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Whole-cell microtiter plate screening assay for terminal hydroxylation of fatty acids by P450s†
Martin J.
Weissenborn‡
a,
Sandra
Notonier‡
a,
Sarah-Luise
Lang
a,
Konrad B.
Otte
a,
Susanne
Herter
b,
Nicholas J.
Turner
b,
Sabine L.
Flitsch
b and
Bernhard
Hauer
*a
aInstitute of Technical Biochemistry, Universitaet Stuttgart, Allmandring 31, 70569 Stuttgart, Germany. E-mail: bernhard.hauer@itb.uni-stuttgart.de
bSchool of Chemistry, University of Manchester, Manchester Institute of Biotechnology, 131 Princess Street, M1 7DN Manchester, UK
First published on 7th April 2016
Abstract
A readily available galactose oxidase (GOase) variant was used to develop a whole cell screening assay. This endpoint detection system was applied in a proof-of-concept approach by screening a focussed mutant library. This led to the discovery of the thus far most active P450 Marinobacter aquaeolei mutant catalysing the terminal hydroxylation of fatty acids.
Directed evolution techniques enable the development of tailor-made biocatalysts exhibiting enhanced catalytic activities, stabilities and substrate selectivities.1 Enzyme engineering requires sensitive, simple to implement and reliable systems of detection.2 Employing evolution methods as a tool to generate large libraries of variants necessitates a smart screening strategy and selection methodology, respectively.3 Accordingly, the system applied for the detection of the desired enzyme activities should be highly sensitive but concomitantly relatively unresponsive to side-reactions and, in particular, applicable in the range of micromolar substrate concentrations.4
Cytochrome P450 monooxygenases (CYPs or P450s) are remarkable enzymes catalysing a broad variety of reactions under mild conditions.5 Previous and successful efforts to engineer P450s focussed on the improvement and variation of activity, chemo-, regio-, and stereoselectivity as well as the catalysis of unnatural carbene reactions.6–9
The reducing equivalents of P450s are mostly provided by the cofactor NAD(P)H. Therefore, monitoring the NAD(P)H consumption as a measure of P450 activity in presence of a substrate seems to be a valid and convenient detection method.10 However, previous work has shown that this technique can yield misleading results (‘false positives’) due to uncoupling events – the consumption of NAD(P)H without the formation of hydroxylated product.2 Even with high coupling efficiencies do NAD(P)H depletion assays suffer from high background signals and hence are not applicable to the more rapid solid-phase assays.11 Also detection methods relying upon the use of alcohol dehydrogenases, which oxidise the hydroxylated compound of interest into the corresponding carbonyl product thereby producing NAD(P)H, are limited due to the aforementioned reasons.12 The background activity with other alcohol dehydrogenases is another factor which has been addressed by the design of artificial cofactors for other enzyme systems recently.13,14
Alternative and rather indirect methods for the detection of P450 activities are based on the use of unnatural substrates to generate colorimetric or fluorescent signals. For instance, indole which spontaneously forms the insoluble dye indigo after P450-catalysed hydroxylation has been successfully applied to the screening for new variants of P450cam from Pseudomonas putida and P450BM3 from Bacillus megaterium (Scheme 1a).15,16 In order to use a substrate which is more similar to the actual substrate of interest, Schwaneberg et al. developed a very elegant strategy to screen and to directly monitor the hydroxylation of aliphatic compounds by employing various fatty acids with terminal p-nitrophenol (PNP) ether moieties (p-nitrophenoxycarboxylic acids).3,17,18 The chromophore PNP gets released upon P450 catalysed hydroxylation of the PNP-binding carbon (Scheme 1b, top). A similar principle was applied in the so-called Purpald® assay.19 Here a methyl ether derivative is employed as a substrate-analogue. The P450 catalysed hydroxylation of the methoxide group results in the formation of formaldehyde (Scheme 1b, bottom). The formaldehyde then reacts with the Purpald® reagent and forms a dark purple colour.
 | ||
| Scheme 1 Comparison of different P450 assays with the herein established detection methodology. | ||
All of these assays, however, rely on substrate-analogues. These analogues are different to the actual substrate of interest. Following the paradigm of Arnold et al. – you get what you screen for – it would be of great use to avoid variations of the substrate in an activity assay intended for the identification of new enzyme variants and activities.20 Comparing a P450 substrate dodecanoic acid (C12) with the substrate-analogue for the purpald assay – decanoic-acid-methylether – reveals two major differences with respect to the enzymatic hydroxylation: (i) the additional ability of the methoxide to accept hydrogen bonds and (ii) the lowered stability of the C–H bond of the methoxide for homolytic cleavage by approximately 6 kcal mol−1 (correlated from ethane for C12 and dimethylether for the decanoic acid methylether).21
Another obstacle in the screening process of P450s is the variation in enzyme expression rates which can lead to ‘false negatives’. The variation in the concentration of active protein between different expression trials is especially significant for P450s and presumably reasoned by their cell toxic activities.22 In this context, an evaluation of the expression level of each P450 variant prior to the activity measurement would be highly useful to find promiscuous and more active mutants.23,24
In the present study, we were interested in developing an assay for the terminal fatty acid hydroxylase CYP153A from Marinobacter aquaeolei in whole cells (use of CYP153AM.aq-CPRBM3).25–28 The assay relies on the substrate conversion by a readily available galactose oxidase (GOase) mutant (Scheme 1c). It is applicable in whole cell P450 transformations for the screening of terminal fatty acid hydroxylases and has accuracy in the micromolar range thereby using the actual substrate of interest (Fig. 1). Implementing a whole cell P450-CO assay – which detects the P450 concentration in whole cells – allowed correlating “cell-activity” to the concentration of active P450 proteins. This enabled the identification of active variants based on the protein concentration and not only based on the efficiency of substrate conversion. The assay reported herein is accurate and sensitive and shortens the otherwise laborious and time intensive fatty acid analysis by a factor of seven.
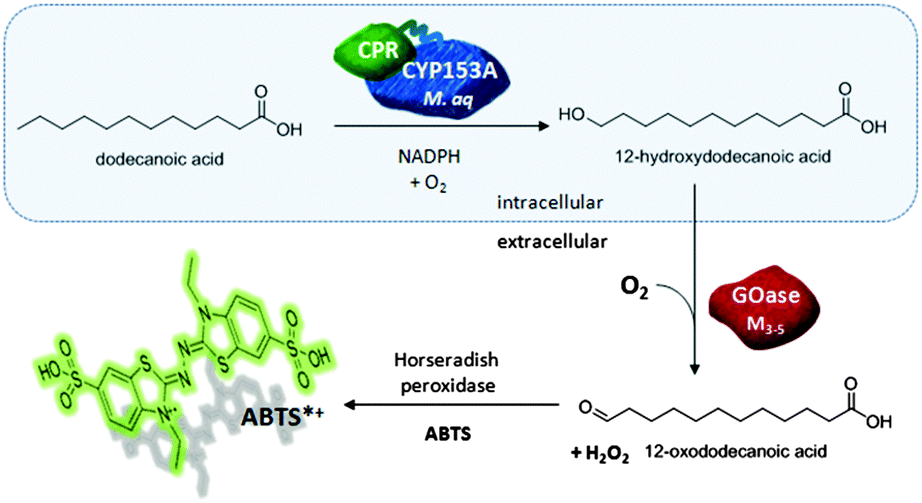 | ||
| Fig. 1 The developed P450 assay: ω-OHC12 formation by CYP153AM.aq-CPRBM3 in whole cells followed by the addition of GOase to the reaction supernatant. GOase is able to oxidize the primary alcohol to the aldehyde which results in the formation of H2O2 as a by-product. The H2O2 can be detected in situ by the common horseradish peroxidase ABTS assay. | ||
We started our investigations into a direct P450 assay by studying different oxidases for the cofactor independent oxidation of alcohols on our example substrate 12-hydroxydodecanoic acid (ω-OHC12). In order to design an assay which is broadly applicable in any laboratory, we focussed on commercially available oxidases: (i) glucose oxidase, (ii) alcohol oxidase from Pichia pastoris and (iii) GOase from Dactylium dendroides. However these oxidases showed no activity towards ω-OHC12 and other aliphatic alcohols (data not shown). We therefore employed the evolved galactose oxidase variant GOaseM3-5 which has been previously shown to possess a wide substrate scope including primary and secondary alcohols.29 With this variant in hand, we tested a range of different alcohols and hydroxylated fatty acids (ω-OHFA) with medium chain length via the commonly used horseradish peroxidase (HRP) ABTS assay and were pleased to find GOaseM3-5 to be active towards all of the compounds tested (Table S2, ESI†). The enzyme displayed higher activity towards primary alcohols when compared to ω-OHFA, whereas the best activity was determined for 1-hexanol while ω-OHC6 was only poorly oxidised.
Encouraged by the results from the activity screen, we next focussed on ω-OHC12 as an example substrate intended to develop a P450 activity assay. The terminal hydroxylation of fatty acids is of industrial relevance and since the analysis of fatty acids by gas chromatography (GC-FID) requires an extraction and additional derivatisation step there is a high demand for a quick and quantitative assay (Fig. 2). The assay was performed using resting cells expressing CYP153AM.aq-CPRBM3 and incubated with 2 mM C12 at 25 °C for 2 h. The biotransformation reaction was terminated by centrifugation yielding a cell-free supernatant followed by the addition of the GOaseM3-5 enzyme in combination with HRP and ABTS, which resulted in a typical colour formation (ABTSox) and an increase in absorbance at λ = 420 nm. However, the change in absorbance could only be observed after several hours, depending on the amount of product formed. This GOase-related lag phase is known, but so far not fully elucidated.30 We hypothesised that the GOase can be inhibited by either an unstable inhibitor or that residual metabolic activity consumed the oxygen required for the GOase reaction. We therefore implemented a heat deactivation step to the procedure after the whole cell reaction and prior to the GOase addition. The supernatant containing the fatty acid product was heated for 30 min at 90 °C. We were able to circumvent the lag phase this way and gained an instant increase of absorption upon GOase addition (Fig. S1, ESI†).
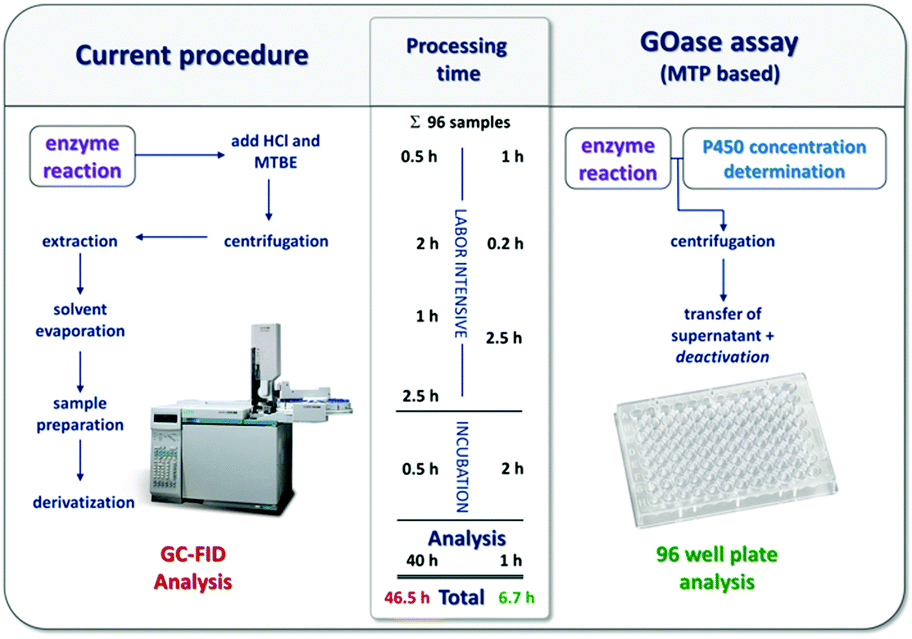 | ||
| Fig. 2 The analysis of fatty acids by gas chromatography and by the GOase based assay. | ||
The change in absorption over time correlated well with product formation of the whole cell P450 reaction as validated by parallel analysis of the product formation by GC-FID (Fig. 3). The detection limit was found to be in the range of 10–20 μM of ω-OHC12. Similar experiments with higher concentrations of ω-OHC12 added to the resting cells confirmed the correlation between the intensity of the signal in the GOase assay and the amount of product being formed (Fig. S2, ESI†). Further controls included the absence of GOase, HRP and ABTS, respectively (Fig. S3, ESI†). The selectivity of the assay for ω-OHC12 was shown by performing the reaction with P450BM3 in place of CYP153AM.aq-CPRBM3. P450BM3 is known to hydroxylate fatty acids in ω-1, ω-2 and ω-3 position.31 By performing the GOase assay with the supernatant of the P450BM3 whole cell reaction, no increase in absorption above the background level could be observed even though product formation was evident by GC-FID analysis (Fig. S4, ESI†). This confirms the selectivity of the assay for terminally hydroxylated substrates.
 | ||
| Fig. 3 Plot representing the production of ω-OHC12 after whole-cell biotransformation and via GOase assay assessment beside GC-FID analysis. | ||
Apart from the determination of P450 activity, we also designed the assay for the evaluation of the expression of active P450 protein. The expression of P450s in E. coli very often suffers from large variations and irreproducibility, partially evoked by inclusion bodies.22 Previous work has shown that the P450 concentration can be assessed via a CO binding assay in whole cells.24 By incubating the whole cells with sodium dithionite for 30 min followed by the addition of CO and incubation for 1 h, we were able to apply this method in microtiter plates (MTPs). A combination of the CO binding assay with the GOase assay enabled us to determine the activity of the P450s and thus to correlate these to the enzyme concentration. Both steps are applicable to MTPs and do not require cell lysis.
Determining not only the enzyme activity, but also its concentration in whole cells was then validated in a proof of concept approach. A focussed P450 mutant library was generated after the creation of a homology model and docking studies and tested for improved activities towards C12 (Table 1). The variants were compared to the so far most active CYP153A mutant G307A.26,32 Three positions within the active site, previously shown to be substrate-interacting, and three positions at the substrate entrance tunnel were selected for mutations (Table S3, ESI†). The substituted residues were chosen based on amino acid frequencies after sequence alignments of the P450 families as described elsewhere.33,34 The library mutants were expressed in a 2 mL final volume in 24 deep-well plates. The cell material from each well was split in two 1 mL-parts: one was screened via the GOase assay for terminal hydroxylation activity and the other was treated with CO to determine the P450 concentration. In order to be able to compare the product concentrations formed and validate the MTP-assay, the reactions were additionally analysed by GC-FID (Table 1 and Table S4, ESI†). To be able to compare the results within different systems and mutants, we set the P450 G307A mutant results to 100% for the MTP-assay and the GC-FID analysis. The results obtained with mutants were calculated relative to the G307A conversion (relative conversion). Judging from the obtained relative conversions, no improved CYP153A variant was found. However, by parallel analysis of the P450 concentration, it was noticed that variant S453A had only a concentration of 0.8 μM whereas the G307A variant showed a concentration of 1.3 μM. Calculating the relative specific conversion – which includes the enzyme concentration – resulted in a 19% more active mutant S453A. These results with the novel most active CYP153A could be confirmed by GC-FID.
| Mutants | Mutation locations | Rel. conversion MTP-assay [%] | P450 conc. [μM] | Rel. specific conversion MTP-assay [%] | Rel. specific conversion GC-FID [%] | Specific activity [μM min−1 μM−1] |
|---|---|---|---|---|---|---|
| G307A | AS | 100 | 1.3 ± 0.03 | 100 | 100 | 2.62 ± 0.57 |
| V306I | AS | 87 ± 0.03 | 1.5 ± 0.22 | 69 ± 0.04 | 62 ± 0.13 | 1.65 ± 1.12 |
| G307R | AS | 0 | 0 | 0 | 0 | 0 |
| F455V | AS | 0 | 0.9 ± 0.21 | 0 | 0 | 0 |
| D134V | SE | 85 ± 0.01 | 1.4 ± 0.02 | 67 ± 0.01 | 45 ± 0.04 | 1.17 ± 0.32 |
| I145L | SE | 82 ± 0.02 | 1.3 ± 0.10 | 65 ± 0.01 | 51 ± 0.08 | 1.34 ± 0.66 |
| S453A | SE | 86 ± 0.02 | 0.8 ± 0.14 | 119 ± 0.03 | 116 ± 0.03 | 3.06 ± 0.28 |
| Empty pET28a(+) | — | — | 0 | 0 | 0 | 0 |
In conclusion, a new microtiter plate-based P450 assay has been developed which utilises the exact substrate of interest and a previously reported GOase variant. The assay has been validated for a range of different substrates and was applied to a focussed mutant library. By implementing an additional CO assay to the work flow the inherent expression problem of P450s could be taken into account to avoid false negatives. The presented assay is quantitative and applicable for small, medium and large mutant libraries. By comparing the current extraction and GC-FID protocol applied to the analysis of fatty acids and related derivatives with the herein newly developed microtiter plate assay, an economy of time is evident as 96 samples can be screened in 2.5 rather than 28 h (Fig. 2).
SN received funding from the People Programme (Marie Curie Actions) of the European Union's 7th Framework Programme (FP7/2007–2013) ITN P4FIFTY under REA Grant Agreement 289217. We also wish to thank Łukasz Gricman (University of Stuttgart) for his help in the generation of the small focussed mutant library and Jens Schmid for the initial experiments. NJT and SLF thank the Royal Society for a Wolfson Research Merit Award.
Notes and references
- U. T. Bornscheuer and M. Pohl, Curr. Opin. Chem. Biol., 2001, 5, 137–143 CrossRef CAS PubMed.
- T. W. Johannes, R. D. Woodyer and H. Zhao, Enzyme Assays: High-throughput Screening, Genetic Selection and Fingerprinting, 2006, pp. 77–93 Search PubMed.
- U. Schwaneberg, C. Otey, P. C. Cirino, E. Farinas and F. H. Arnold, J. Biomol. Screening, 2001, 6, 111–117 CAS.
- M. Alcalde, E. T. Farinas and F. H. Arnold, J. Biomol. Screening, 2004, 9, 141–146 CrossRef CAS PubMed.
- R. Bernhardt, J. Biotechnol., 2006, 124, 128–145 CrossRef CAS PubMed.
- R. Bernhardt and V. B. Urlacher, Appl. Microbiol. Biotechnol., 2014, 98, 6185–6203 CrossRef CAS PubMed.
- O. Salazar, P. C. Cirino and F. H. Arnold, ChemBioChem, 2003, 4, 891–893 CrossRef CAS PubMed.
- T. S. Wong, F. H. Arnold and U. Schwaneberg, Biotechnol. Bioeng., 2004, 85, 351–358 CrossRef CAS PubMed.
- P. S. Coelho, E. M. Brustad, A. Kannan and F. H. Arnold, Science, 2013, 339, 307–310 CrossRef CAS PubMed.
- G. E. Tsotsou, a. E. G. Cass and G. Gilardi, Biosens. Bioelectron., 2002, 17, 119–131 CrossRef CAS PubMed.
- S. J. Park and J. R. Cochran, Protein Eng. Des., 2009, 146 CAS.
- Y. Yang, J. Liu and Z. Li, Angew. Chem., Int. Ed., 2014, 53, 3120–3124 CrossRef CAS PubMed.
- C. E. Paul, S. Gargiulo, D. J. Opperman, I. Lavandera, V. Gotor-Fernández, V. Gotor, A. Taglieber, I. W. C. E. Arends and F. Hollmann, Org. Lett., 2013, 15, 180–183 CrossRef CAS PubMed.
- S. A. Löw, I. M. Löw, M. J. Weissenborn and B. Hauer, ChemCatChem, 2016, 8, 911–915 CrossRef.
- Q. S. Li, U. Schwaneberg, P. Fischer and R. D. Schmid, Chem. – Eur. J., 2000, 6, 1531–1536 CAS.
- A. Çelik, R. E. Speight and N. J. Turner, Chem. Commun., 2005, 3652–3654 RSC.
- U. Schwaneberg, C. Schmidt-Dannert, J. Schmitt and R. D. Schmid, Anal. Biochem., 1999, 269, 359–366 CrossRef CAS PubMed.
- A. Glieder, E. T. Farinas and F. H. Arnold, Nat. Biotechnol., 2002, 20, 1135–1139 CrossRef CAS PubMed.
- P. Meinhold, M. W. Peters, A. Hartwick, A. R. Hernandez and F. H. Arnold, Adv. Synth. Catal., 2006, 348, 763–772 CrossRef CAS.
- C. Schmidt-Dannert and F. H. Arnold, Trends Biotechnol., 1999, 17, 135–136 CrossRef CAS PubMed.
- F. Agapito, B. J. Costa Cabral and J. a. Martinho Simões, THEOCHEM, 2005, 719, 109–114 CrossRef CAS.
- M. Lindmeyer, D. Meyer, D. Kuhn, B. Bühler and A. Schmid, J. Ind. Microbiol. Biotechnol., 2015, 42, 851–866 CrossRef CAS PubMed.
- S.-J. Choi, M. Kim, S.-I. Kim and J.-K. Jeon, J. Biochem. Mol. Biol., 2003, 36, 332–335 CrossRef CAS PubMed.
- W. A. Johnston, W. Huang, J. J. De Voss, M. A. Hayes and E. M. J. Gillam, J. Biomol. Screening, 2008, 13, 135–141 CrossRef CAS PubMed.
- D. Scheps, S. H. Malca, H. Hoffmann, B. M. Nestl and B. Hauer, Org. Biomol. Chem., 2011, 9, 6727–6733 CAS.
- S. Honda Malca, D. Scheps, L. Kühnel, E. Venegas-Venegas, A. Seifert, B. Nestl and B. Hauer, Chem. Commun., 2012, 48, 5115–5117 RSC.
- S. M. Hoffmann, M. J. Weissenborn, Ł. Gricman, S. Notonier, J. Pleiss and B. Hauer, ChemCatChem, 2016 DOI:10.1002/cctc.201501397.
- M. T. Lundemo, S. Notonier, G. Striedner, B. Hauer and J. M. Woodley, Appl. Microbiol. Biotechnol., 2016, 100, 1197–1208 CrossRef CAS PubMed.
- F. Escalettes and N. J. Turner, ChemBioChem, 2008, 9, 857–860 CrossRef CAS PubMed.
- T. S. Moon, D. R. Nielsen and K. L. J. Prather, AIChE J., 2012, 58, 2303–2308 CrossRef CAS.
- L. Narhi and A. Fulco, J. Biol. Chem., 1986, 261, 7160–7169 CAS.
- D. Scheps, S. Honda Malca, S. M. Richter, K. Marisch, B. M. Nestl and B. Hauer, Microb. Biotechnol., 2013, 6, 694–707 CAS.
- Ł. Gricman, C. Vogel and J. Pleiss, Proteins: Struct., Funct., Bioinf., 2014, 82, 491–504 CrossRef PubMed.
- Ł. Gricman, C. Vogel and J. Pleiss, Proteins, 2015, 83, 1593–1603 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, additional tables and figures. See DOI: 10.1039/c6cc01749e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |