
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Glycomimetic-based pharmacological chaperones for lysosomal storage disorders: lessons from Gaucher, GM1-gangliosidosis and Fabry diseases
Elena M.
Sánchez-Fernández
a,
José M.
García Fernández
*b and
Carmen Ortiz
Mellet
 *a
*a
aDepartamento de Química Orgánica, Facultad de Química, Universidad de Sevilla, Profesor García González 1, 41012, Sevilla, Spain. E-mail: mellet@us.es
bInstituto de Investigaciones Químicas (IIQ), CSIC – Universidad de Sevilla, Avda. Américo Vespucio 49, 41092 Sevilla, Spain. E-mail: jogarcia@iiq.csic.es
First published on 22nd March 2016
Abstract
Lysosomal storage disorders (LSDs) are often caused by mutations that destabilize native folding and impair the trafficking of enzymes, leading to premature endoplasmic reticulum (ER)-associated degradation, deficiencies of specific hydrolytic functions and aberrant storage of metabolites in the lysosomes. Enzyme replacement therapy (ERT) and substrate reduction therapy (SRT) are available for a few of these conditions, but most remain orphan. A main difficulty is that virtually all LSDs involve neurological decline and neither proteins nor the current SRT drugs can cross the blood–brain barrier. Twenty years ago a new therapeutic paradigm better suited for neuropathic LSDs was launched, namely pharmacological chaperone (PC) therapy. PCs are small molecules capable of binding to the mutant protein at the ER, inducing proper folding, restoring trafficking and increasing enzyme activity and substrate processing in the lysosome. In many LSDs the mutated protein is a glycosidase and the accumulated substrate is an oligo- or polysaccharide or a glycoconjugate, e.g. a glycosphingolipid. Although it might appear counterintuitive, substrate analogues (glycomimetics) behaving as competitive glycosidase inhibitors are good candidates to perform PC tasks. The advancements in the knowledge of the molecular basis of LSDs, including enzyme structures, binding modes, trafficking pathways and substrate processing mechanisms, have been put forward to optimize PC selectivity and efficacy. Moreover, the chemical versatility of glycomimetics and the variety of structures at hand allow simultaneous optimization of chaperone and pharmacokinetic properties. In this Feature Article we review the advancements made in this field in the last few years and the future outlook through the lessons taught by three archetypical LSDs: Gaucher disease, GM1-gangliosidosis and Fabry disease.
 Elena M. Sánchez-Fernández | Elena M. Sánchez-Fernández received her PhD degree in 2006 at the University of Granada (Spain). Then she moved to the Institute for Chemical Research (IIQ, Seville) funded with a three-year postdoctoral contract from the Spanish National Research Council (CSIC). In 2010 she got a Marie Curie Fellowship at the Chemistry Research Laboratory, University of Oxford (UK). Since 2013 she has been working as a postdoctoral Marie Curie Reintegration Grant Fellow at the University of Seville. Her main interests focus on the synthesis of glycodrugs and their applications in therapies for lysosomal storage diseases, cancer, infection and inflammation. |
 José M. García Fernández | Jose Manuel García Fernández received his PhD degree from the University of Seville (Spain) in 1988. Between 1990 and 1995 he pursued postdoctoral research at the Centre d'Etudes de Grenoble (France) with Dr Jacques Defaye. In 1996 he joined the Spanish National Research Council (CSIC) at the Institute for Chemical Research (IIQ), where he currently serves as a Research Professor and Director. He has authored more than 200 scientific articles and he is a co-inventor of 17 patents. Current targets of the laboratory include the control of carbohydrate–protein and carbohydrate–nucleic acid interactions with glycomimetics for applications in nanomedicine, gene delivery, cancer and protein folding diseases. |
 Carmen Ortiz Mellet | Carmen Ortiz Mellet received her PhD degree in Chemistry from the University of Seville (Spain) in 1984, where she was appointed Tenure Professor of Organic Chemistry in 1987. In 1990 and 1995 she joined the group of Jacques Defaye (Centre d'Etudes de Grenoble, France) to work on the synthesis of complex thiooligosaccharides and cyclodextrins. Since 1998 she has been responsible for the Carbohydrate Bioorganic Chemistry Group, being promoted to Full Professor in 2008. Ongoing projects include the development of glycomimetic-based therapies for lysosomal storage disorders and cancer and the design of self-assembled glycomaterials for drug and gene delivery. |
Introduction
Lysosomal storage disorders (LSDs) are a clinically heterogeneous group of more than 40 inherited orphan conditions, with an overall prevalence estimated to be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1500–1:7000 live births, sharing a common pathobiochemical leitmotiv: a genetic defect in the genes that encode specific lysosomal enzymes leading to the storage of complex non-metabolized molecules in the lysosome.1 Substrate accumulation then results in chronic and progressive clinical syndromes that often display a wide spectrum of abnormalities that are unique to each LSD. Consequently, the primary target of any therapeutic strategy towards LSDs, whether approved or experimental, is restoring the balance between substrate influx and degradation in the key cells and tissues.2 In a large proportion of cases, the lysosomal enzymes at the origin of these diseases are soluble acidic glycosidases produced in the endoplasmic reticulum (ER), quality checked, and transported to the lysosome via the Golgi apparatus.3,4 The origin of protein dysfunction causing LSDs is diverse; however, abnormal protein folding during biosynthesis in the ER is often observed. Enzyme replacement therapy (ERT),5 in which patients are regularly supplemented with an exogenous recombinant enzyme, and small-molecule substrate reduction therapy (SRT),6,7 relying on the inhibition of substrate biosynthesis, represent the primary treatment options that are approved for patients with some LSDs. More recently, an alternative therapeutic paradigm consisting of the elaboration of specific ligands that selectively bind and stabilize otherwise unstable mutant enzymes to increase total cellular levels and improve lysosomal trafficking and activity, so-called pharmacological chaperones (PCs), has emerged (Fig. 1).8,9
:1500–1:7000 live births, sharing a common pathobiochemical leitmotiv: a genetic defect in the genes that encode specific lysosomal enzymes leading to the storage of complex non-metabolized molecules in the lysosome.1 Substrate accumulation then results in chronic and progressive clinical syndromes that often display a wide spectrum of abnormalities that are unique to each LSD. Consequently, the primary target of any therapeutic strategy towards LSDs, whether approved or experimental, is restoring the balance between substrate influx and degradation in the key cells and tissues.2 In a large proportion of cases, the lysosomal enzymes at the origin of these diseases are soluble acidic glycosidases produced in the endoplasmic reticulum (ER), quality checked, and transported to the lysosome via the Golgi apparatus.3,4 The origin of protein dysfunction causing LSDs is diverse; however, abnormal protein folding during biosynthesis in the ER is often observed. Enzyme replacement therapy (ERT),5 in which patients are regularly supplemented with an exogenous recombinant enzyme, and small-molecule substrate reduction therapy (SRT),6,7 relying on the inhibition of substrate biosynthesis, represent the primary treatment options that are approved for patients with some LSDs. More recently, an alternative therapeutic paradigm consisting of the elaboration of specific ligands that selectively bind and stabilize otherwise unstable mutant enzymes to increase total cellular levels and improve lysosomal trafficking and activity, so-called pharmacological chaperones (PCs), has emerged (Fig. 1).8,9
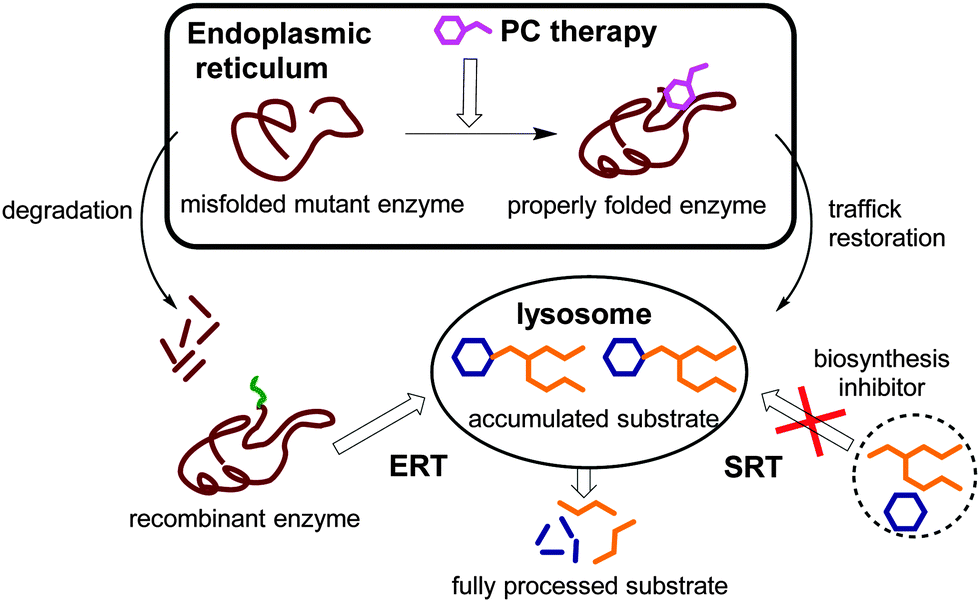 | ||
| Fig. 1 Schematic representation of the therapeutic strategies available for LSDs: ERT, enzyme replacement therapy; SRT, substrate reduction therapy; and PC, pharmacological chaperone. | ||
PCs are low molecular weight compounds that stabilize the native conformation of a mutant enzyme in the ER, allowing it to escape aggregation and premature degradation by the ER-associated degradation pathway. The properly folded mutant enzyme, stabilized by the PC, can then be transported to the Golgi apparatus for maturation and reach the lysosome, increasing the residual enzyme activity of the cells. Although it might appear contradictory, most PCs have also been competitive inhibitors of their target enzyme.10 Once the PC:enzyme complex reaches the lysosome, the large amounts of stored substrates are believed to displace the PC and take over the stabilization of the mutant enzyme. Contrary to ERT and SRT, PC therapy addresses not only substrate accumulation but also the protein folding defects and their potential contributions to the pathophysiology of the disease.11 Moreover, PCs can in principle be subjected to medicinal chemistry optimization strategies to improve pharmacokinetics, toxicity and biodistribution properties, particularly in view of reaching the central nervous system.12 This is of special relevance to LSDs, since most of these conditions imply neurological symptoms that cannot be addressed by ERT strategies.13
Some molecules with therapeutic promise as PCs have been identified by high throughput screening of libraries in view of drug repositioning.14 In such cases there is no apparent resemblance between the structure of the chaperone and the substrate, which in the case of a lysosomal glycosidase is an oligosaccharide or a glycoconjugate. Lately, macromolecular chaperone candidates consisting of multiple copies of a sugar-like motif displayed at the surface of a polyantennaeted scaffold have also been proposed.15,16 The focus of this Feature Article, however, is glycomimetic-based active site-directed pharmacological chaperones: small molecules that emulate the structure of the monosaccharide unit cleaved off by the target LSD-associated glycosidase, eventually incorporating additional non-glycone moieties intended to improve mutant enzyme binding, folding and rescuing capabilities.17 Glycomimetics can be designed to achieve all of the requirements for a potent therapeutic drug and thereby create a new and innovative source of novel therapeutics.18 Understanding the detailed molecular basis of the interaction between the native or mutant enzyme and the natural sugar substrate or their functional analogues is essential for these channels. X-ray crystallography of the co-crystallized glycomimetic ligand and the lysosomal glycosidase provide the most reliable data for PC rational design.19 This knowledge is complemented by the evolving understanding of the mechanisms leading to loss of function of mutant forms that retain catalytic activity or that have only modestly compromised function due to premature degradation or ER aggregation.20 To illustrate the current developments in glycomimetic-based PC design, we have selected relevant examples, preferentially from contributions made in the last five years that involve either in cellulo or in vivo evaluation, for three representative LSDs: Gaucher disease (GD), GM1-gangliosidosis (GM1) and Fabry disease (FD).
GD is an inherited metabolic disorder caused by mutations in the glucocerebrosidase gene (GBA1) that results in defective and insufficient activity of the enzyme β-glucocerebrosidase (EC 3.2.1.45; GCase), which catalyzes the hydrolysis of the β-glycopyranosyl linkage in glucosylceramide (1; Fig. 2).21 For operational purposes, GD can be subdivided into 3 types based on the age at onset and neurological manifestations. Type 1 GD was considered to have no discernible neuronopathic features. The infantile-onset acute neuronopathic form, Type 2, carries a very severe prognosis, whereas Type 3 GD is a chronic neuronopathic form. Although GD is the most common LSD, it is nevertheless relatively rare and classified as an orphan disease. The prevalence of GD in the general population is estimated to be 1 in 57000 live births, but this increases to 1 in 1000 in Ashkenazi Jews, with 5–10% of patients worldwide having the neuronopathic forms. Whereas ERT and SRT are available for Type 1 GD, to date, there are no approved treatments for the neurological manifestations of Type 2 and 3 GD.22 Recent findings have also drawn attention to impaired cognition, albeit subtle, in Type 1 GD patients, a manifestation that is not addressed by ERT or SRT. The frequency of GD and Lewy-body dementia is also greatly increased in the healthy parents and other heterozygous GD carriers. Additionally, mutations in the GBA1 gene confer the greatest risk of Parkinsonism in all populations.23
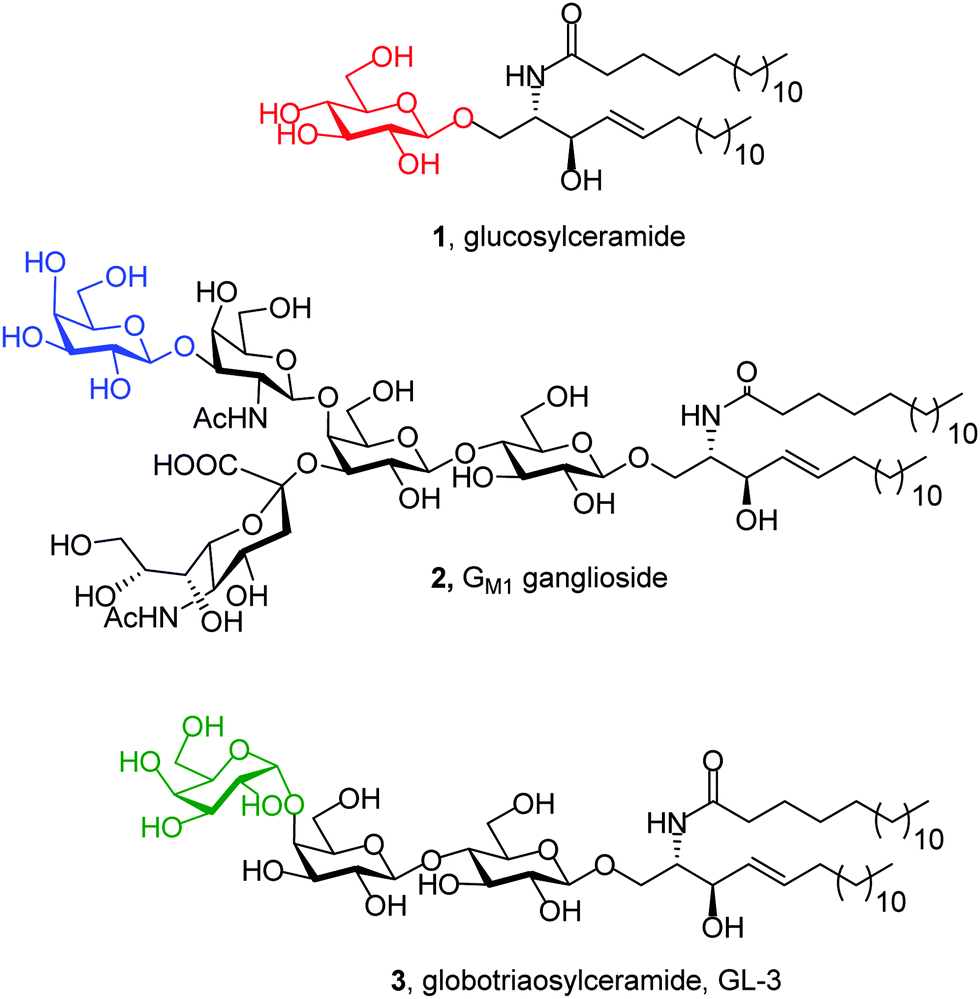 | ||
| Fig. 2 Structures of the substrates accumulated in the lysosomes of patients suffering from Gaucher disease (1), GM1 gangliosidosis (2) and Fabry disease (3). The monosaccharide unit cleaved off by the corresponding disease-associated lysosomal glycosidase is colored in red, blue and green, respectively. | ||
Mutations in the GLB1 gene encoding for the precursor form of human lysosomal β-galactosidase (EC 3.2.1.23; β-Gal) are at the origin of GM1, an autosomal, recessive, severe neurodegenerative disorder that has been classified into three clinical phenotypes, infantile, juvenile, and adult forms, depending on the onset and severity.24 β-Gal catalyzes the hydrolysis of terminal β-galactopyranosyl residues from various substrates, including ganglioside GM1 (2; Fig. 2). The estimated incidence of GM1 is 1:100000–200000 live births, with no effective medical treatment available; the inability of proteins to cross the blood–brain barrier makes ERT inefficient when the central nervous system is involved.25
FD is an X-linked LSD caused by mutations in the GLA gene that encodes lysosomal α-galactosidase A (EC 3.2.1.22; α-Gal A). α-Gal A cleaves α-linked galactopyranosyl moieties of neutral glycosphingolipids, mainly globotriaosylceramide (3, GL-3; Fig. 2). Deficiency in α-Gal A activity results in the accumulation of GL-3, which gives rise to a variety of clinical manifestations such as cardiomyopathy, renal dysfunction, stroke, and, in some cases, neurological symptoms. Although FD follows X-linked inheritance, heterozygous females can be symptomatic. The incidence of FD, according to the literature, varies from 1:40000 to 1:170000 live births. While ERT is available, its impact on the life quality of patients is not always satisfactory.26 Indeed, ERT in advanced FD seems to be of doubtful benefit and improved treatment options are needed.27
While all these three diseases originate from the arrest of glycosphingolipid metabolism, the critical biochemical step at which it blocks is different for each condition. Reactivation of substrate hydrolysis with pharmacological chaperones implies rescuing the precise dysfunctional glycosidase without interfering with any other enzymatic process. Selectivity is going to be, thus, a critical aspect for clinical prospects that must be considered in any glycomimetic chaperone design. It entails endowing the candidates with configurational (e.g., between enzymes acting on gluco or galacto substrates) and anomeric (e.g., between enzymes acting on β- or α-galactopyranosyl substrates) discrimination capabilities. Mimicking the configurational pattern of the glycone part of the putative substrate with iminosugars, monosaccharide analogues in which the endocyclic oxygen is replaced by an amine nitrogen, has been by far the most thoroughly investigated approach. Indeed, iminosugars had already met considerable success in the discovery and elaboration of competitive glycosidase inhibitors.28–30 Tuning chaperoning properties turned to be a more complex task, however, requiring higher doses of creativity eventually leading to new PC prototypes. Efforts towards new PC therapies for Gaucher disease, the LSD that has preferentially attracted the chemists' attention, better illustrate this point. For the sake of clarity, we have chosen to subdivide the discussion of this section by compound families rather than following a chronological scheme. The sections regarding GM1 and FD serve to highlight the importance of moulding glycomimetic structure to impart selectivity towards enzymes differing on the anomeric configuration of the hydrolysed glycosidic bond. Opportunities for further improving chaperone function by chemical tailoring will be presented in the Concluding remarks and future outlook section.
Glycomimetic-based PCs for Gaucher disease
Unmodified iminosugars
The focus for pharmacological chaperone candidates was initially placed at unmodified iminosugars behaving as competitive inhibitors of the target lysosomal enzyme, i.e. β-glucocerebrosidase in the case of Gaucher disease. Among those, isofagomine (IFG, 4) is the compound that reached a more advanced stage in view of drug development (planned trademark Plicera®). IFG exhibited promising results ex vivo and in vivo as a GCase enhancer,31 with several mutant GCases associated with GD showing increases in enzymatic activity and protein in response to the chaperone. Deceivingly, IFG failed to proceed to Phase II clinical trials (Phase III in combined therapy with recombinant GCase) after scarce improvement in key GD markers.32,33In vivo studies with a mouse model for neuronopathic Gaucher disease showed that IFG significantly attenuated the progression of the disease by increasing GCase levels and mediating the suppression of proinflammation, but this was not correlated with a reduction in the accumulation of lipid substrates.34,35 One caveat is that IFG is an inhibitor of GCase in the lysosome. Although this inhibitory effect is reversible, a net gain in enzyme activity is only achieved after the removal of the drug from the medium, meaning that medical use would probably require a very delicate intermittent dosing of the iminosugar. It is also of note that it requires a three orders of magnitude higher concentrations of IFG to increase GCase activity in cells (10–100 μM) compared with the amount needed to inhibit GCase in the in vitro assays (5–100 nM).36 The most likely explanation for this difference is that IFG, a highly hydrophilic molecule, is transported into the ER and the lysosome very inefficiently; similar unfavourable membrane-crossing properties may apply for other canonic iminosugars evaluated as pharmacological chaperones for GD such as calystegines A3 (5), B1 (6), B2 (7) and C1 (8), 1,5-dideoxy-1,5-iminoxylitol (DIX, 9),37,38 noeurostegine39 (10) or the 3,4,5,6-tetrahydroxyazepane 11 (Fig. 3).40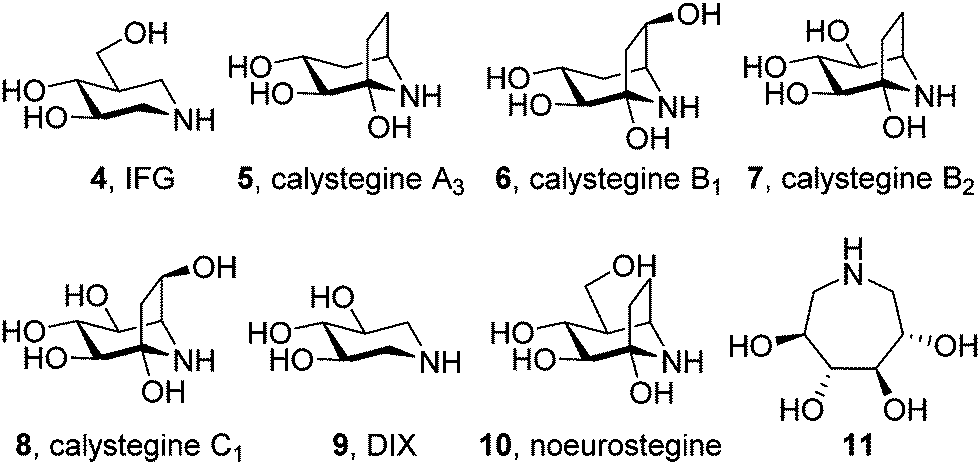 | ||
| Fig. 3 Structures of some unmodified iminosugars behaving as GCase inhibitors and chaperones. | ||
N-Alkylated iminosugars
The discovery that iminosugars with N-linked alkyl chains of varying lengths, e.g. N-butyl and N-nonyl DNJ (NB-DNJ and NN-DNJ; 12 and 13, respectively; Fig. 4), can bind to GCase and stabilize substrate bound conformations of mutant GCase forms had a considerable impact in the field by providing an appealing strategy to improve the metabolic properties of chaperone aspirants (cell and ER permeability, for example). The alkyl chains of NB-DNJ and NN-DNJ are oriented towards the entrance of the active site in the corresponding chaperone:GCase complexes, participating in favourable hydrophobic contacts with amino acid residues, contributing to the complex stability and ensuring the correct initial folding and trafficking of mutant GCase to the lysosome.41 For the particular case of the N370S mutation, the most prevalent in Type 1 GD patients, structural and biochemical data indicated that the protein is already correctly folded and that the mechanism by which competitive inhibitors increase the lysosomal levels of GCase is by reducing degradation by proteases within the lysosome rather than by having an effect on protein folding and trafficking.42 NB-DNJ, and possibly other chaperones, also mediates the formation of the enzyme–substrate complex in the lysosome and the interaction of GCase with saponin C,43 an established activator for the hydrolysis of glucosylceramide by GCase in lysosomes that is compromised in the N370S mutant.44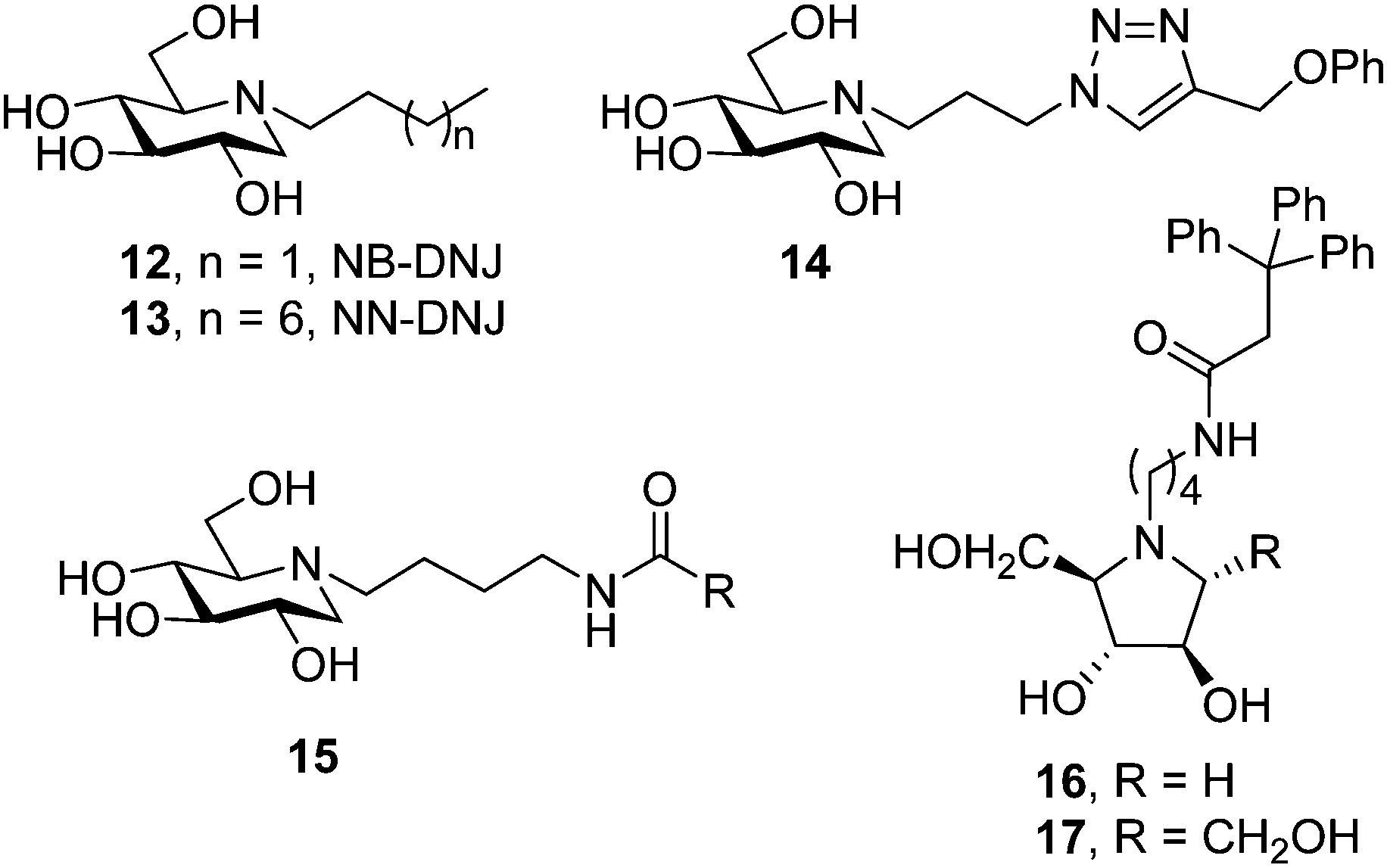 | ||
| Fig. 4 Structures of some N-alkylated iminosugars evaluated as PCs for GD. | ||
Following the pioneering work on NB-DNJ and NN-DNJ, several examples of N-alkylated iminosugar frameworks have been proposed as pharmacological chaperones for GD. The common concept is to conjugate an iminosugar with a lipophilic moiety; the former may mimic the sugar part or the transition state towards glycosidic cleavage and the latter may mimic the ceramide aglycone of the natural substrate, glucosylceramide (1, Fig. 2). To accelerate chaperone discovery, molecular diversity strategies have been implemented. Thus, in 2011 Gouin et al.45 developed an efficient “click” procedure to tether hydrophobic substituents to DNJ through the popular copper catalyzed azide–alkyne cycloaddition (CuAAC) reaction.46 Cycloadduct 14, bearing a phenoxymethylene group at position C-4 of the triazol ring (Fig. 4), was identified as the most promising compound from a set of fourteen amphiphilic DNJ derivatives, increasing mutant GCase activity in N370S/N370S Gaucher cells nearly 2-fold at 20 μM compared to untreated cells. In the same optics, Cheng et al.47 used N-(4-aminobutyl) derivatives of DNJ, 2,5-dideoxy-2,5-imino-D-arabinitol (DAB) and 2,5-dideoxy-2,5-imino-D-mannitol (DMDP) as scaffolds to generate a large library of glycomimetics by amidation of the terminal primary amine group in the appendage. The authors identified potent GCase inhibitors in the three series, but whereas DNJ candidates (15) did also inhibit lysosomal α-glucosidase, the pyrrolidine counterparts were selective for the target enzyme, with compounds 16 and 17 (Fig. 4) promoting 2.5- and 2.2-fold activity enhancements in homozygous N370S human fibroblasts at 6.2 and 1.5 μM concentrations, respectively.
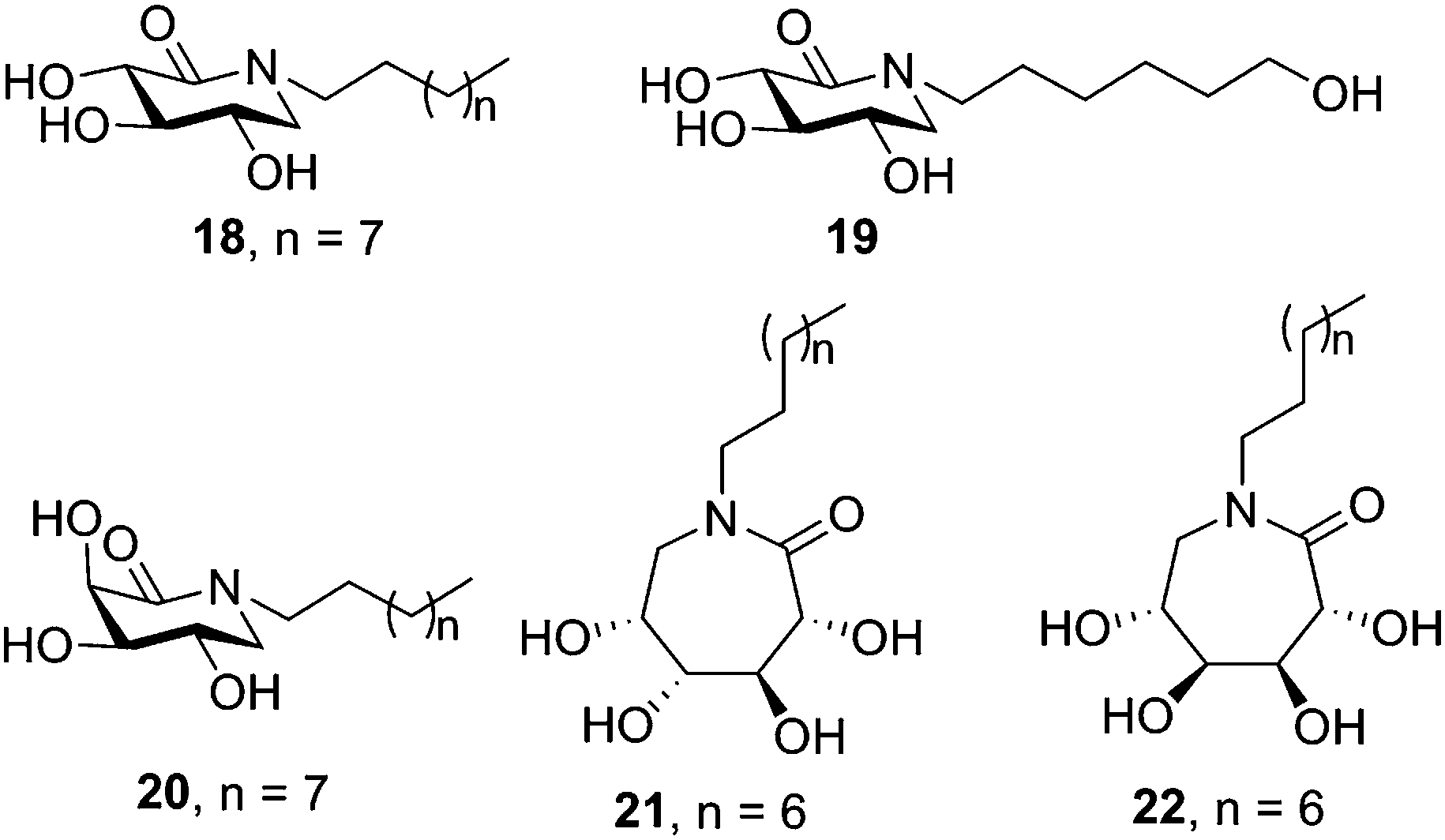
Butters, Ye and coworkers proposed a different prototype for GD chaperones based on N-substituted δ-lactams with a carbonyl group instead of the hydroxymethyl group in a deoxynojirimycin (DNJ) scaffold.48 The sp2-hybridized carbon was designed to distort the ring to a half-chair conformation, which according to docking experiments was expected to lead the protruding carbonyl group to interact with the surrounding amino acids and stabilize the substrate-bound conformation of GCase. Indeed, several of the synthesized N-alkylated derivatives with a configurational pattern matching that of DNJ (D-gluco) elicited significant enzyme activity enhancements in Gaucher lymphoblasts (N370S), with the N-octyl and N-(6-hydroxyhexyl) derivatives 18 and 19 (50 μM concentration; Fig. 5) reaching outstanding 6.2- and 4.1-fold relative to untreated control cells, respectively. It is worth noting that δ-lactams related to the galacto-type epimer of DNJ (D-galacto configuration) also showed GCase chaperoning activity in the same GD cells (e.g. 2.9-fold for the N-decyl derivative 20 at 50 μM). Further work by the same group showed that the homologous ε-glucono- and ε-galactono-lactams (e.g. the N-nonyl derivatives 21 and 22, respectively; Fig. 5) did also increase GCase activity in N370/N370 GD lymphoblasts up to 2.4 fold at 50 μM concentration, meaning a better performance as compared with NB-DNJ and NN-DNJ in spite of their neutral character.49
 | ||
| Fig. 5 Structures of N-alkylated iminosugar lactams behaving as GD PCs. | ||
C-Alkylated iminosugars
Isofagomine-type iminosugar glycomimetics (1-azasugars), which comprise IFG, the calystegines and DIX (Fig. 3), bind to GCase with the nitrogen atom occupying a portion analogous to the anomeric carbon of glucose in GCase-bound glucosylceramide (C-1), whereas deoxynojirimycin-type iminosugars do so in a mode in which the nitrogen atom matches the endocyclic oxygen (O-5). The ensemble of data on N-alkylated iminosugars as GD pharmacological chaperones support that in the second binding mode accommodation of the N-substituent in the hydrophobic pocket of GCase is very favourable, to the point that it imparts GCase inhibition properties even when the parent iminosugar may not be an inhibitor of the enzyme. In stark contrast, N-alkylation of iminosugar behaving as 1-azasugars, for instance the N-octyl derivative 23 (Fig. 6), is either detrimental or has little effect on GCase binding affinity.50 Interestingly, moving the alkyl substituent from the N-atom to the adjacent C-atom in 1-azasugars restores the glycomimetic:GCase fitting capabilities, leading to strong GCase inhibitors51 with GCase chaperoning properties, such as the C-6-propyl IFG derivative 24 (1.5-fold GCase activity enhancement in N370S GD fibroblasts).52 The potential of this strategy to develop new GD chaperones was elegantly demonstrated by Compain, Martin and Asano, who developed α-1-C-nonyl-DIX (25; Fig. 6) as one of the most potent mutant GCase enhancers ever reported: 1.8-fold activity increase in N370S fibroblasts at a concentration as low as 10 nM.53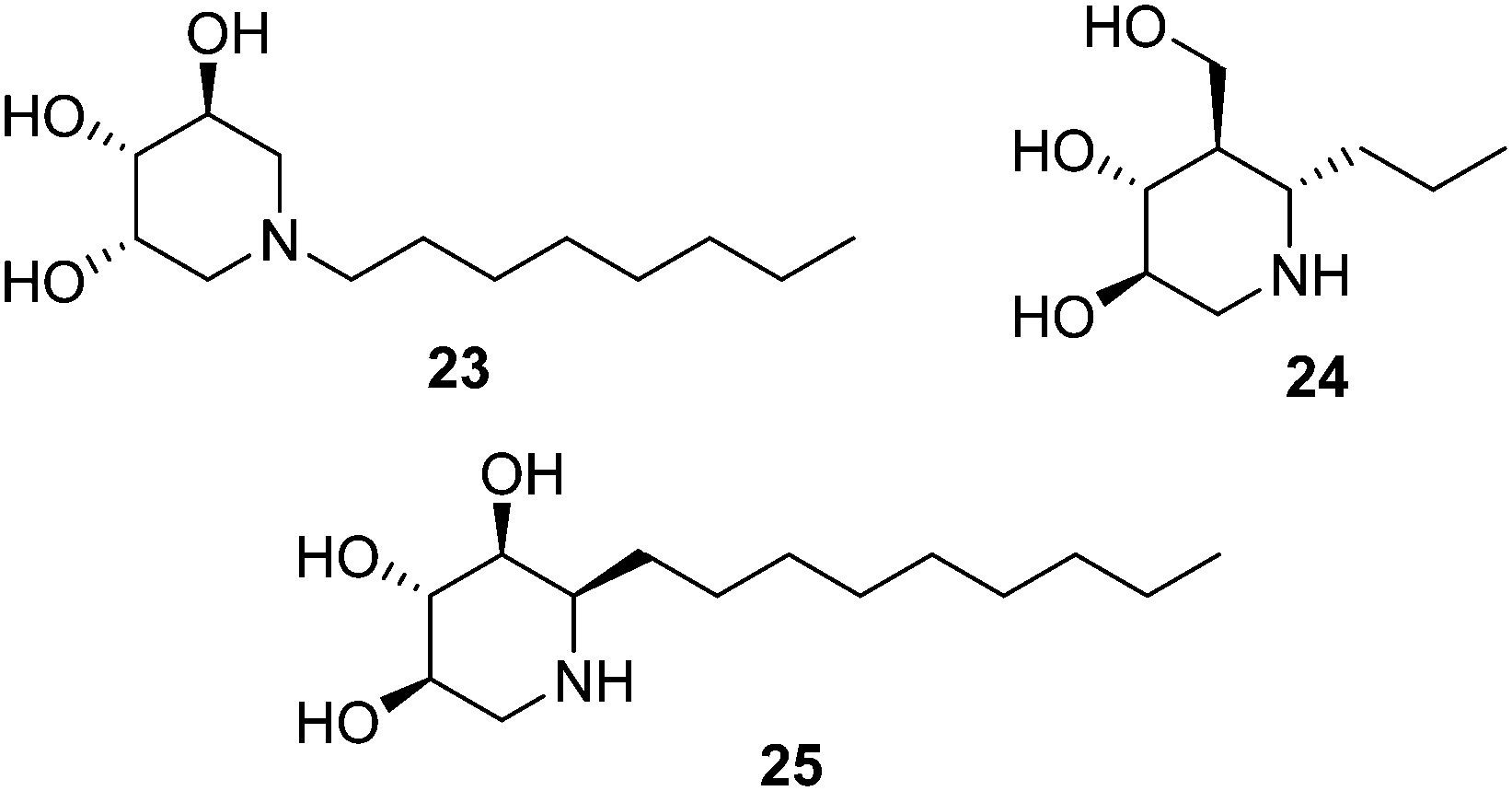 | ||
| Fig. 6 Structure of the N- and C-alkylated 1-azasugar derivatives 23–25. | ||
Withers and coworkers took advantage of the thiol–ene click-type reaction54 to rapidly generate a collection of sixteen 1-C-alkylated DIX derivatives bearing various lipophilic substituents (general structure 27) from the unsaturated DIX precursor 26 and different thiols (Fig. 7, upper lines).55 Library members 28 and 29 exhibited the highest mutant GCase activity enhancement capabilities among the prepared compounds, reaching 3.4- and 1.4-fold in homozygous Gaucher N370S and L444P fibroblasts, respectively, when used at 100 μM concentration. The latter result is particularly notable, given that the L444P mutation, with a high prevalence in Type 3 Gaucher patients,56 is not located in the GCase domain harbouring the catalytic site57 and is refractory to most pharmacological chaperone candidates (Fig. 8).
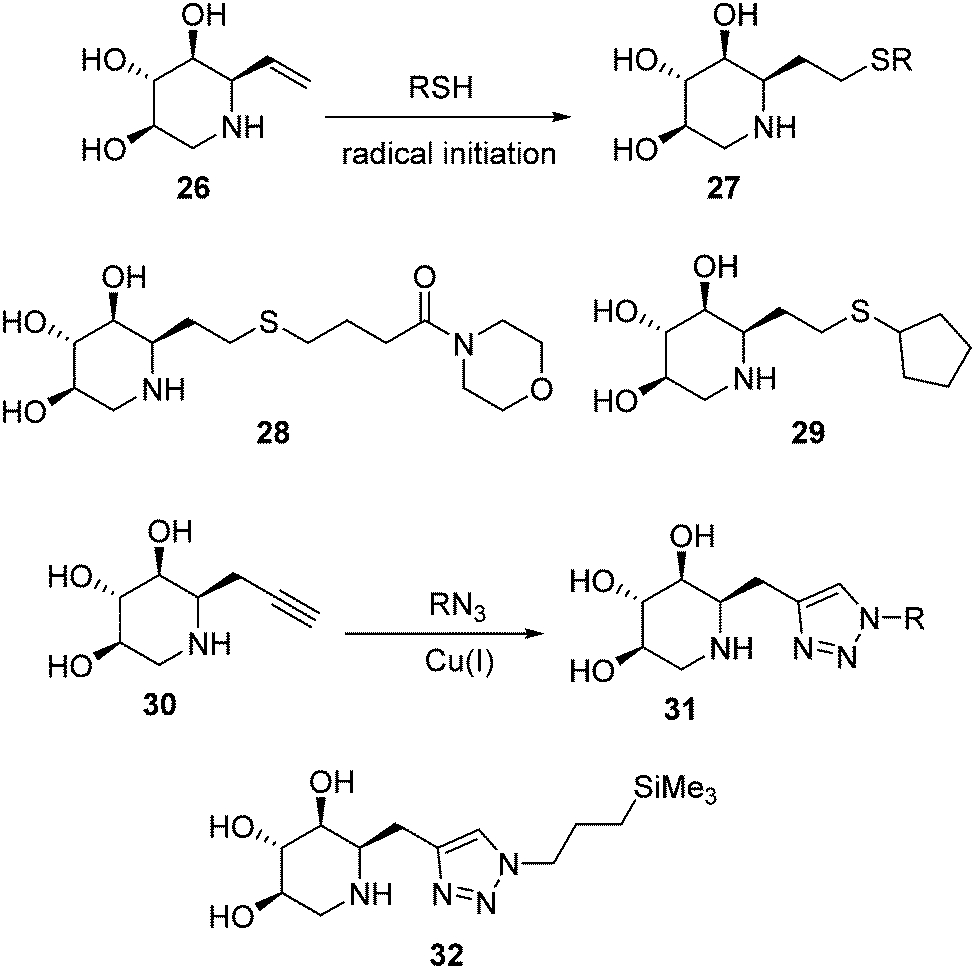 | ||
| Fig. 7 Thiol–ene (upper lines) and CuAAC (lower lines) strategies for the generation of C-alkylated DIX derivatives as PCs for GD. | ||
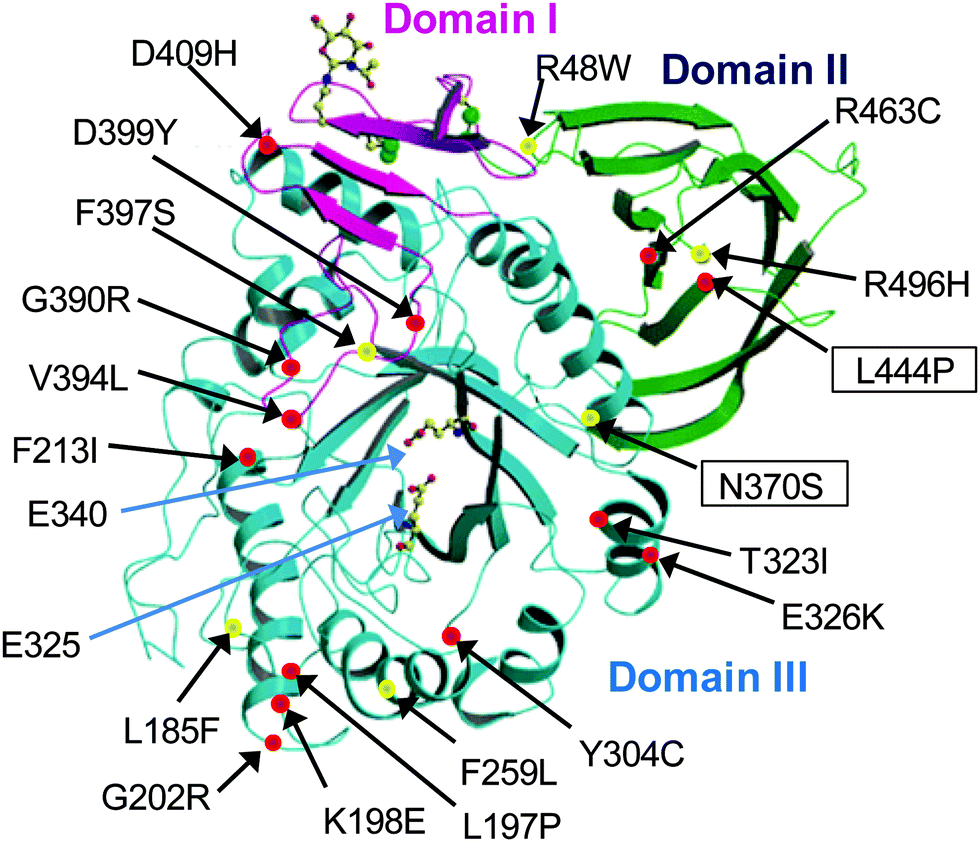 | ||
| Fig. 8 X-ray structure of acid-β-glucosidase (reproduced with permission from ref. 57; ©2003 European Molecular Biology Organization). Domain I is shown in magenta, domain II in green and domain III, which is the catalytic domain, in blue. The active-site residues E235 and E340 are shown as ball-and-stick models. Some representative GD-associated GCase mutations are shown as balls, with those that cause predisposition to severe (types 2 and 3) and mild (type 1) disease in red and yellow, respectively. The most common GCase mutations in non-neuronopathic (N370S) and neuronopathic (L444P) forms of GD are shown in boxes. | ||
More recently, Compain, Delgado and coworkers expanded the battery of 1-C-alkylated DIX derivatives by using the alkynyl DIX precursor 30 and applying CuAAC ligation chemistry, thereby accessing 1,2,3-triazole adducts 31 (Fig. 7, lower lines).58 The 3-(trimethylsilyl)propyl derivative 32, exhibited the best activity enhancement within the prepared compounds, reaching up to 4-fold in Gaucher fibroblasts containing the G202R mutation in homo or heterozygosis, associated with the neuronopathic phenotype of the disease (Fig. 8), at 100 nM concentration.
Amphiphilic pyrrolidine-type iminosugars can probably adapt to either of the two above commented glycomimetic orientations, namely with the nitrogen atom in a location equivalent to C-1 or O-5 in glucopyranosides, upon binding to GCase, depending on whether the lipophilic substituent is attached to the nitrogen atom (e.g.16 and 17) or to an adjacent carbon atom. For instance, the α-1-C-tridecyl-DAB derivative 33 (Fig. 9) reported by Kato and coworkers59 behaved as a strong, selective and competitive inhibitor of GCase (Ki 0.71 μM) and was able to enhance the activity of the mutant enzyme in N370S/N370S Gaucher fibroblasts by 1.5-fold at 1 μM concentration, meaning a ten times lower optimal concentration as compared with isofagomine (4, Fig. 3). A docking study of 33 to human GCase supported the 1-azasugar binding mode, benefitting from a hydrogen bonding network involving the protonated imino group and the triol system analogous to that encountered for IFG and from favourable hydrophobic contacts between the tridecyl chain and the amino acids coating the hydrophobic pocket at the entrance of the catalytic site.
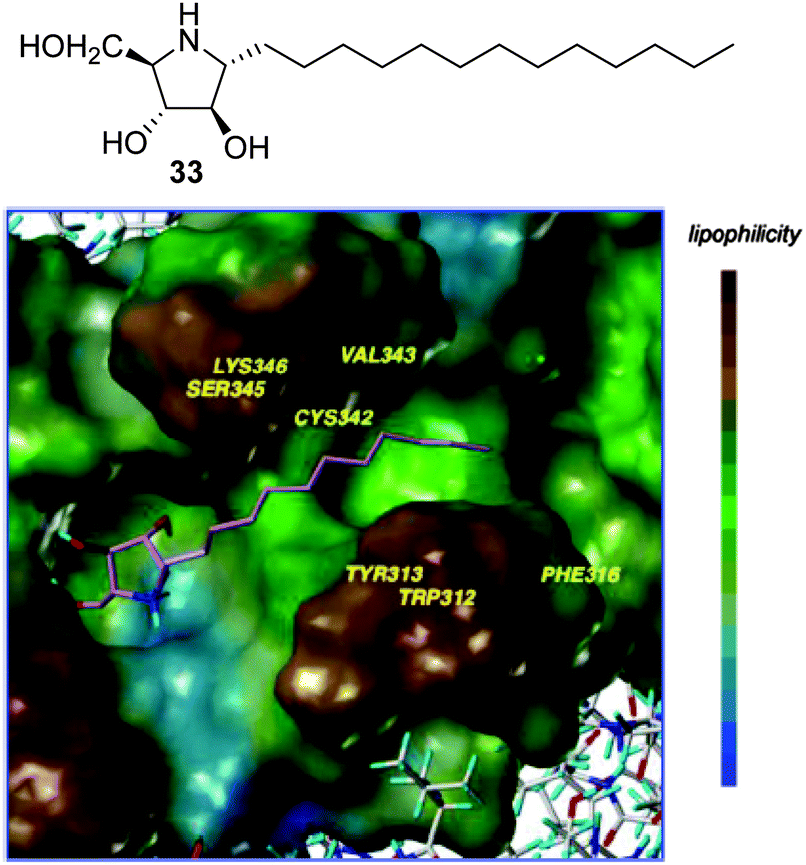 | ||
| Fig. 9 Structure of α-1-C-tridecyl-DAB (33) and its binding in the active site of GCase as obtained by docking. | ||
sp2-Iminosugars
The chemical reactivity and conformational properties of classical iminosugars is overwhelmingly dominated by the amine character of the sp3-hybridized nitrogen atom in the azaheterocyclic skeleton. Most importantly, the intrinsic instability of aminal functionalities makes iminosugars unsuitable to emulate the acetal group of glycosides, the natural substrates of glycosidases. Analogues of N- and S-glycosides, incorporating gem-diamine or aminothioacetal pseudoanomeric centers, are likewise labile, which in practice restrict the possibility to incorporate pseudoaglycon-like moieties with a precise orientation to the synthetically more demanding iminosugar-C-glycoside representatives.60 The failure at properly mimicking the anomeric linkage is probably at the origin of the low selectivity frequently encountered for iminosugars towards enzymes acting on anomeric substrates as well as among isoenzymes, which seriously hampers their progress into the clinics. Moreover, owing to their basic character iminosugars are largely protonated at physiological pH, which further restrains the possibility to convey significant binding affinity differences towards the target glycosidase at neutral (ER) and acidic environments (lysosome), a critical aspect for chaperone candidates.Replacement of the underlining amine group of iminosugars into a trigonal planar pseudoamide-type nitrogen (N-carbonyl, N-thiocarbonyl, N-imino group), with substantial sp2-hybridation nature, drastically modifies the chemical and stereoelectronic properties of iminosugar frameworks, offering broad opportunities for pKa tuning compatible with the incorporation of virtually any type of exocyclic substituent. Members of this family of glycomimetics, generically termed sp2-iminosugars, such as the natural alkaloid kifunensine (34) or the synthetic glycoamidines (e.g.35), glycohydroxymolactams (e.g.36), glycoimidazoles (e.g.37) and glycotetrazoles (e.g.38), have been known for a long time (Fig. 10),61 but the expansion of sp2-iminosugars as glycosidase activity modulators started only in the late 90's with the initial report on the inhibitory properties of hemiaminal-type representatives (Fig. 11).62 The p orbital locating the lone pair of the endocyclic N-atom in pseudoamide functionalities, with π-symmetry, very efficiently overlaps with the σ*-antibonding orbital of an axially-oriented contiguous pseudoanomeric bond, drastically enhancing the hyperconjugative contribution to the generalized anomeric effect, enabling access to unique glycomimetics with stable N–C1–X (X = O, N, S or C) pseudoanomeric centers.63
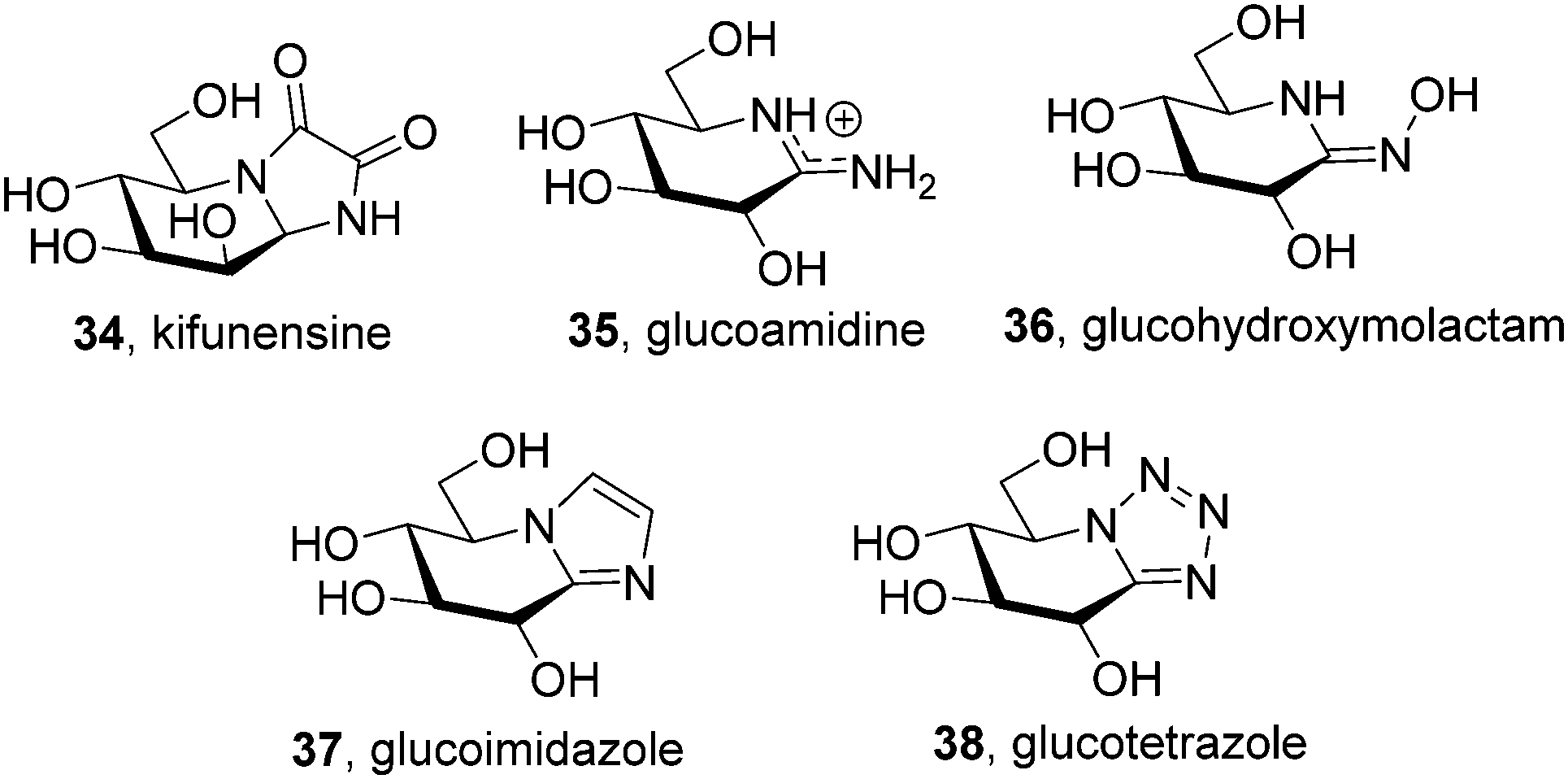 | ||
| Fig. 10 Structure of some natural and synthetic non-reducing sp2-iminosugars. | ||
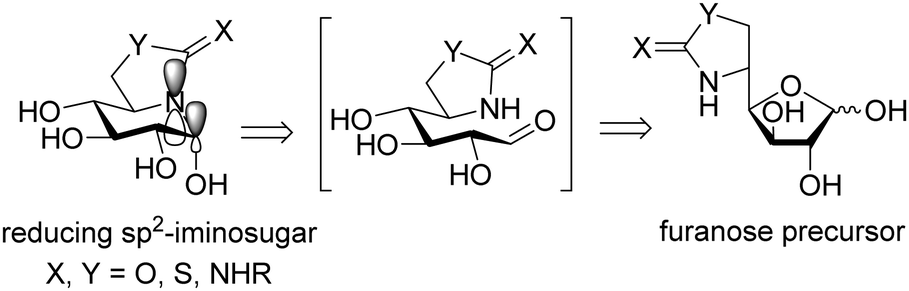 | ||
| Fig. 11 General structure and retrosynthetic scheme for reducing sp2-iminosugars (D-gluco series); the p and σ* orbitals involved in hyperconjugative contribution to the anomeric effect are depicted. | ||
The chemical synthesis of reducing sp2-iminosugars is significantly simplified as compared to classical iminosugars and takes advantage of the chemoselective intramolecular nucleophilic addition of pseudoamide nitrogens (carbamate,64 thiocarbamate,65 urea,66 thiourea,67 isourea,68 isothiourea,69 guanidine,70 sulfamide,71 and thiohydantoine72) to the masked aldehyde group of a carbohydrate template in the open chain form (Fig. 11). The substitution pattern, configurational profile and ring size of the final compounds can be varied in a predictable manner by tailoring the monosaccharide precursor, making the procedure very well-suited for diversity-oriented strategies. Such favorable features have been exploited to optimize the chaperoning capabilities towards several LSD-associated glycosidases.
Amphiphilic bicyclic sp2-iminosugars related to the natural reducing alkaloid nojirimycin (NJ), such as 5N,6O-(N′-octyliminomethylidene)nojirimycin (39, NOI-NJ) or its 6-thio (40, 6S-NOI-NJ) and 6-amino-6-deoxy (41, 6N-NOI-NJ) analogues (Fig. 12A), are anomeric-specific inhibitors of β-glucosidases, including the lysosomal acid β-glucosidase (GCase) associated with Gaucher disease (IC50 values 1.3–14.4 μM).73 X-ray structural evidence demonstrated that the axial orientation of the pseudoanomeric OH group in the ground state of the glycomimetics in aqueous solution switch to equatorial (β-configuration) when complexed with the enzyme, with the octyl chain locating at the hydrophobic pocket at the entrance of the catalytic site (Fig. 12B and C).74 Interestingly, all three compounds behaved as about one-order-of-magnitude stronger inhibitors of GCase at neutral (pH 7) than at acidic pH (pH 5.2) in human normal as well as Gaucher fibroblasts. The chaperoning capabilities were assessed by determining the enzyme activity enhancements in GD cells from patients with seven different GCase mutations, namely F213I/F213I, G202R/L444P, N188S/G193W, N370S/N370S, F213/L444P, L444P/RecNcil and L444P/L444P, in comparison with NN-DNJ (13, Fig. 4). All the sp2-iminosugars proved largely superior to NN-DNJ in the F213I, G202R and N188S variants, associated with orphan neuronopathic forms of the disease, with GCase activity enhancements ranging from 2 to 3-fold at 30 μM. In cellulo ten-day's time course experiments showed that a maximum activity enhancement is achieved at day 3–4, then reaching a plateau with no affectation of cell viabilities. When cells were deprived of the chemical chaperones on day 4, the activity gradually decreased to the basal level within one to three days (Fig. 12D). The hypothesized endogenous GCase rescuing and trafficking restoring mechanism was confirmed by colocalization experiments using a fluorescently labeled sp2-iminosugar chaperone in combination with immunodetection of GCase, calnexin (ER) and the lysosomal-associated membrane protein-2 (LAMP-2).75
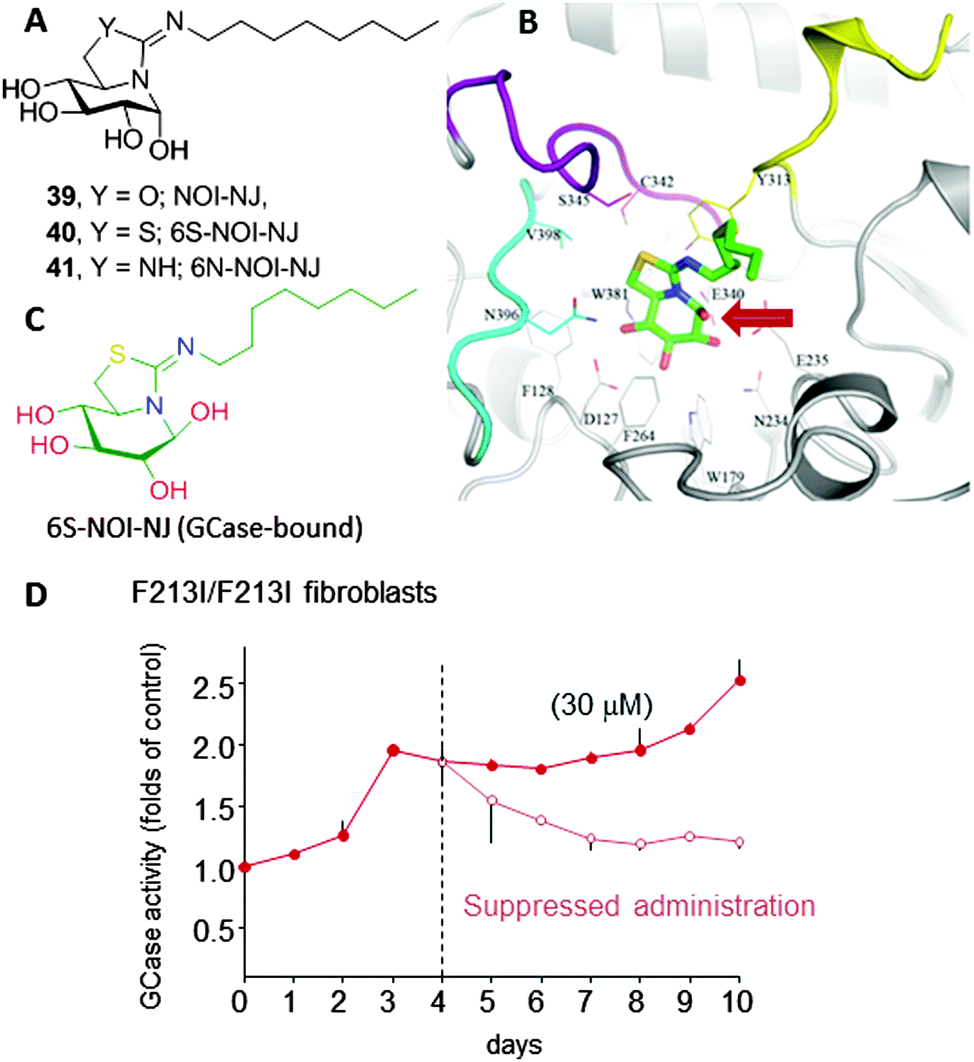 | ||
| Fig. 12 (A) Structures of 5N,6O-(N′-octyliminomethylidene)nojirimycin (39, NOI-NJ), its 6-thio (40, 6S-NOI-NJ) and 6-amino-6-deoxy (41, 6N-NOI-NJ) analogues in water solution; note the α-configuration of the pseudoanomeric center and the axial disposition of the corresponding hydroxyl. (B) Crystal structure of the complex between 6S-NOI-NJ and recombinant human GCase; the anomeric hydroxyl is indicated with a red arrow. (C) Representation of GCase-bound 6S-NOI-NJ showing the β-oriented anomeric OH group. (D) Chaperone activity of 6N-NOI-NJ in homozygous F213I Gaucher fibroblasts (10-days time course; filled circles); a subset of cells was cultured with chaperones for four days, washed and further cultured without the drug for six days (empty circles). | ||
Most in vitro studies focusing on the basic mechanism and small compound screening efforts for LSD therapies have been performed on patient fibroblasts, a cell type not primarily affected in patients. In 2013, Tiscornia et al.76 described the development of an induced pluripotent stem cell (iPSc) model for the acute neuronopathic form of GD (early onset GD Type 2) that showed the same GCase mutations (G202R/L444P) and low GCase activity as the original fibroblasts they were derived from. They subsequently differentiated the iPSc into dopaminergic neurons and used them as a platform to test the chaperone capabilities of a focused library of reducing sp2-iminosugars related to nojirimycin and galactonojirimycin. Two compounds, namely the isourea-type derivative NOI-NJ and the isothiourea analogue 5N,6S-N′-[4-(adamantane-1-caboxamido)butyliminomethylidene]-6-thionojirimycin (6S-AdBI-NJ),77 were found to increase GCase levels and activity in the differentiated neuronal cultures over 3-fold at a relatively low concentration (30 μM; Fig. 13A). Notably, 6S-AdBI-NJ is very well adept at forming inclusion complexes with β-cyclodextrin (βCD) derivatives, which was further exploited to elaborate chaperone delivery systems targeting macrophages, the most affected cells in GD patients, such as the trimannosylated βCD conjugate Man3-βCD (Fig. 13B).78 Given estimates that only small increases in GCase activity would be required to achieve a clinical effect, these results support further development of these compounds as chaperone candidates for Type 2 neuronopathic GD, which currently lacks therapeutic options and is lethal.
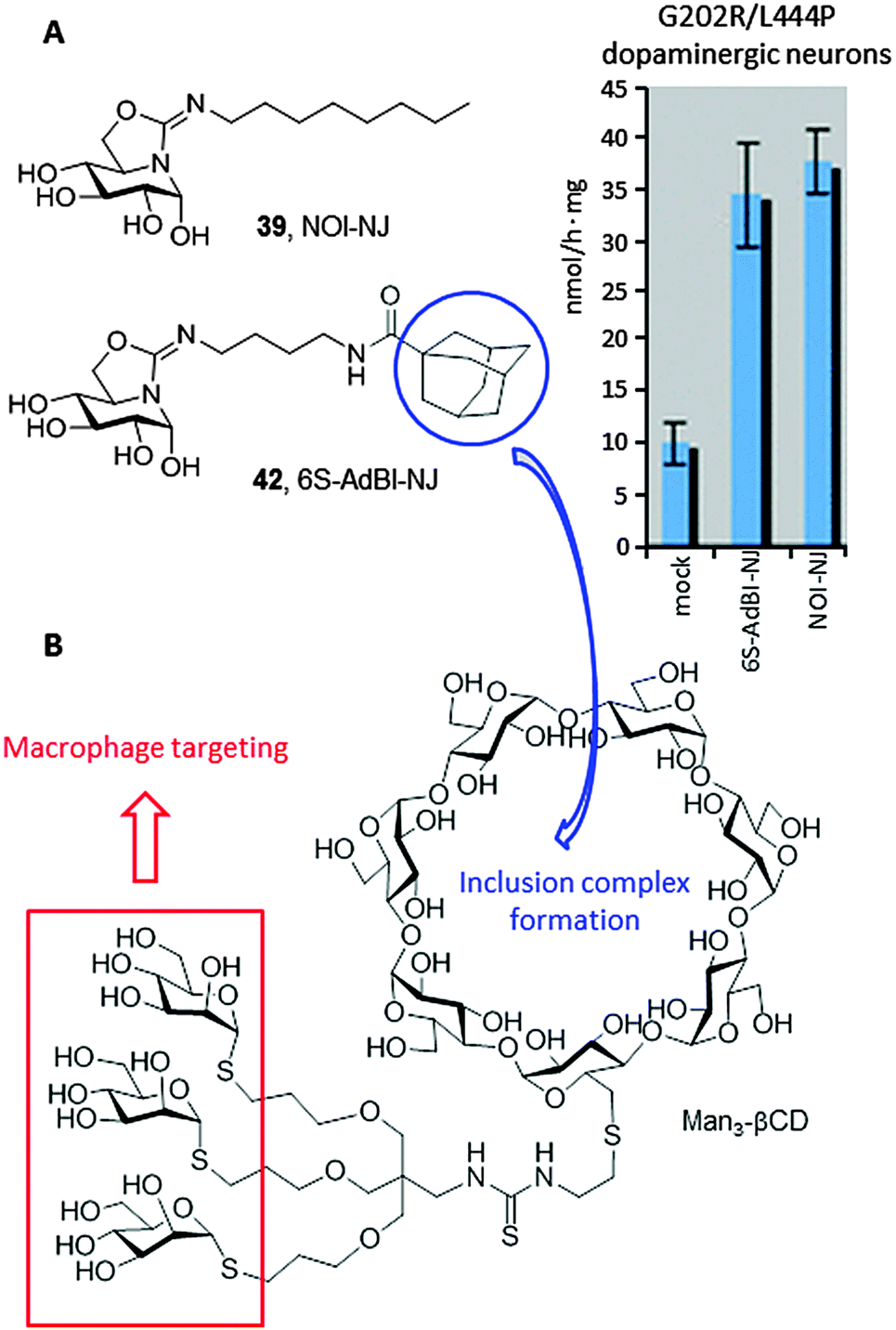 | ||
| Fig. 13 (A) Structures of the sp2-iminosugar chaperones NOI-NJ (39) and 6S-AdBI-NJ (42) and GCase activity enhancements in G202R/L444P dopaminergic neurons differentiated from induced pluripotent stem cells treated with 30 μM concentrations of the chaperones. (B) Structure of the cyclodextrin conjugate Man3-βCD devised for macrophage-selective delivery of 6S-AdBI-NJ. | ||
sp2-Iminosugars having a fused bicyclic skeleton systematically failed to promote significant activity GCase enhancements in cells with the L444P/L444P genotype, one of the most prevalent GD-causing mutations in humans that is associated with the late onset neuronopathic variant (GD Type 3). A survey of crystal structures of chaperone:GCase complexes evidenced that the enzyme has a propensity to induce a conformational change in the azaheterocycle ring of classical79 as well as sp2-iminosugars,74 from chair to envelop or skew-boat, to optimize fitting. This feature seems to restrain the correct folding stabilization and trafficking restoration capabilities of the chaperones to those cases where the point mutation is located in the catalytic domain of GCase. Interestingly, undistortable sp2-iminosugars related to the calystegine iminosugar family,80 such as the N-[(4-adamantanecarboxamido)butylthiocarbamoyl]-1,6-anhydro-L-idonojirimycin derivative 43 (NAdBT-AIJ; Fig. 14A), were found to be effective at propagating the GCase refolding effect in stable transfected COS-7 cells expressing L444P GCase as well as in fibroblasts of GD patients homozygous for this mutation, facilitating translocation of the enzyme from the ER to the lysosome and leading to above 3-fold GCase activity enhancements.81
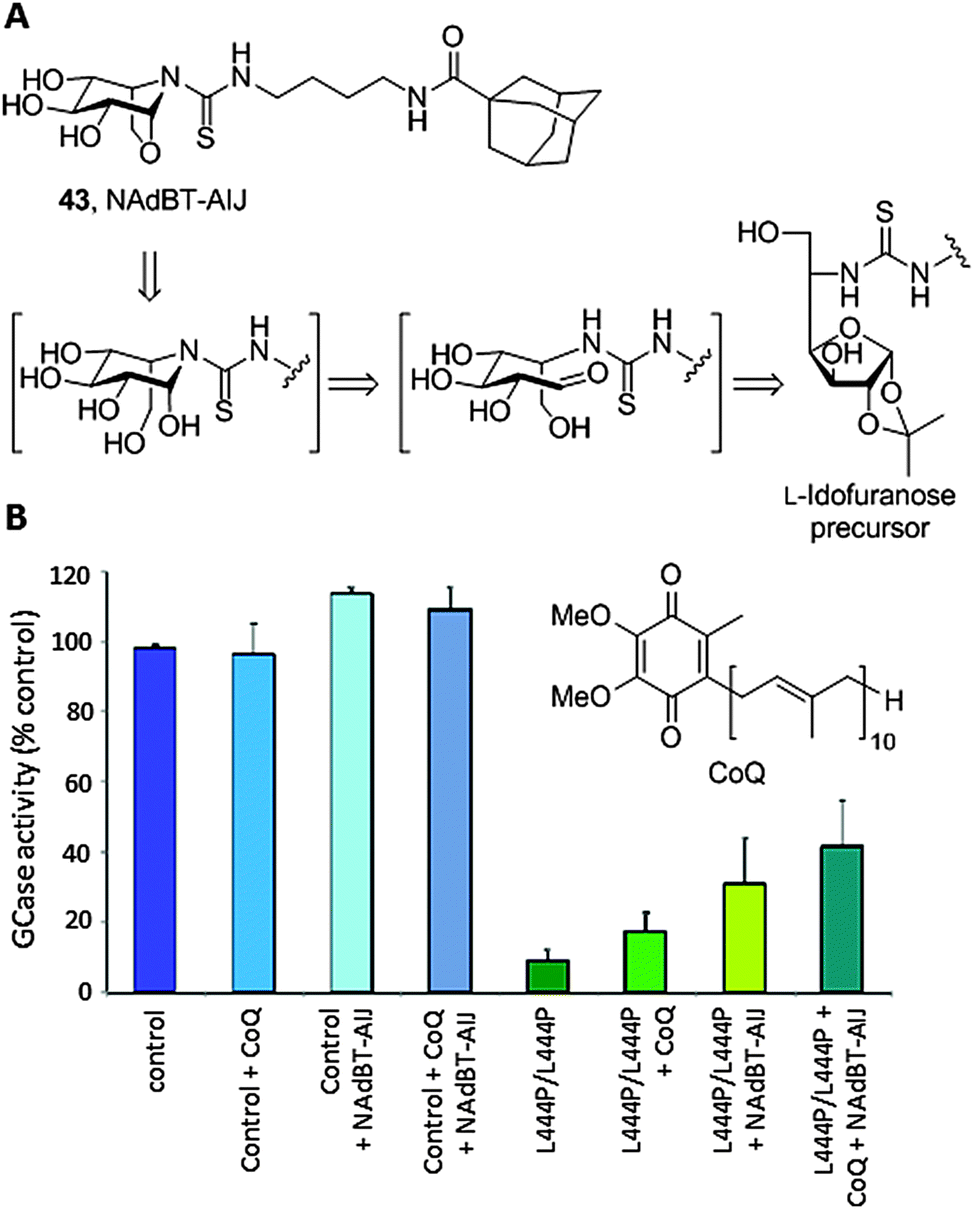 | ||
| Fig. 14 (A) Structure and retrosynthetic scheme for the rigid sp2-iminosugar chaperone NAdBT-AIJ. (B) GCase activity enhancements in normal (control) and L444P/L444P GD fibroblasts promoted by coenzyme Q10 (CoQ, 25 μM), NAdBT-AIJ (25 μM) and the combined treatment with CoQ + NAdBT-AIJ (25 μM + 25 μM). | ||
The oxa-nor-tropane skeleton in NAdBT-AIJ was built from a 5-deoxy-5-thioureido-L-idonofuranose precursor following acid-catalyzed deprotection. Sequential furanose → piperidine rearrangement and intramolecular glycosidation involving OH-6 occurred spontaneously,82 affording the calystegine-type chaperone in high yield (Fig. 14A). The efficiency of NAdBT-AIJ in improving the L444P/L444P GD condition in fibroblasts was further confirmed by Sánchez-Alcazar and coworkers.83 These authors demonstrated that, in addition to the lysosome, the mitochondria function is severely affected in GD patients, leading to membrane depolarization, reduced ATP levels, increase in reactive oxygen species (ROS) production, mitophagy activation and impaired autophagic flux. Based on these findings, they conceived a combined therapy targeting both organelles by co-administration of the NAdBT-AIJ chaperone and coenzyme Q10 (CoQ), an antioxidant and mitochondrial energizer that is currently considered as a potential experimental drug for the treatment of neurodegenerative diseases in general84 and lysosomal diseases in particular.85 Significant synergistic effects were observed, which in addition to the accessibility of both active agents makes the strategy very promising for the treatment of neuronopathic forms of GD that are not responsive to ERT (Fig. 14B).
Aminocyclitols
Most research efforts towards the design of glycomimetic-type pharmacological chaperones have taken advantage of the opportunities that incorporation of nitrogen functionalities in polyhydroxylated scaffolds offers for pH-dependent (therefore cell organelle-dependent) modulation of the affinity and selectivity towards the target enzyme and improvement of the pharmacokinetic properties. Nitrogen-in-the-ring monosaccharide mimics have been generally privileged, but alternative prototypes with the key nitrogen functionality at an exocyclic location, such as the sugar-like aminocyclitols, have also shown considerable promise. Thus, in 2004 Lin et al. reported on the GCase chaperoning capabilities of N-octyl-β-valienamine (NOV, 44; Fig. 15) in GD fibroblasts harbouring the F213I/F213I mutation.86 A computational study supported that NOV (predicted pKa = 8.69) is protonated in the whole pH 5–7 window.87 At pH 7 (ER), the aspartic acid residue Asp127 of GCase is deprotonated and tolerates a deeper binding of the chaperone, allowing an efficient interaction between the amino group of NOV and the catalytic acid Glu235. Protonation of Asp127 at pH 5 (lysosome) weakens the interaction and promotes dissociation of NOV from the enzyme, allowing substrate processing (Fig. 15). The location of the N-atom is thus critical for the NOV mechanism of action and possibly for other aminocyclitols.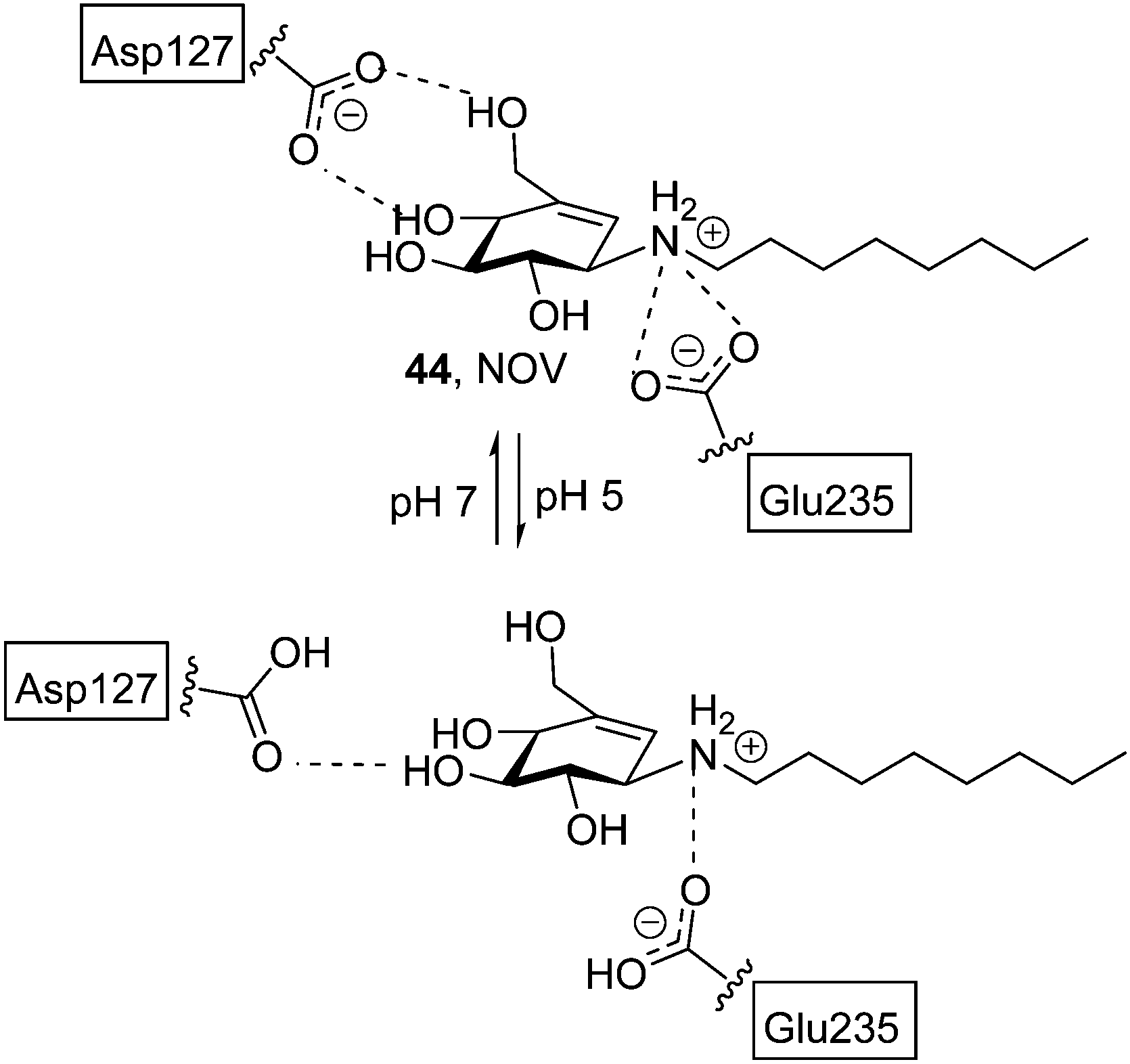 | ||
| Fig. 15 Schematic representation of the interactions of NOV with Asp127 and Glu235 at the active site of GCase at pH 7 (Asp127 deprotonated) or pH 5 (Asp127 protonated) as obtained by computational calculations. | ||
Casas and coworkers and Llebaria and co-workers have conducted an intense research on the synthesis and evaluation of aminocyclitol-type chaperones, with several candidates for GD on record. In 2009 it was found that amino-scyllo-inositol derivative 45 (Fig. 16), bearing an N-decyl substituent, behaved as a pharmacological chaperone in GD patient fibroblasts bearing the neuronopathic L444P/G202R and L444P;E326K/G202R genotypes.88 Two years later bicyclic isoureas and guanidines with cis ring fusion (e.g. amino-myo-inositol derivatives 46 and 47) were developed as extremely potent inhibitors of recombinant GCase (Ki values in the range 2–10 nM).89 The most efficient compound in this work, namely 46, promoted a 2.1-fold GCase activity enhancement in N370S/N370S Gaucher lymphoblasts when used at 100 nM concentration. Remarkably, GCase activity in L444P/L444P lymphoblasts was also increased by up to 50% at 10 nM. Further improvement was achieved by preparing diantennaeted cyclitols bearing two vicinal alkylamino groups (e.g.48)90 or, even better, aminocyclitols with adjacent N- and O-alkyl substituents: compound 49 (Fig. 16), incorporating two nonyl chains, produced maximum increases of GCase activities of 90% in N370S lymphoblasts at 1 nM and 40% in L444P lymphoblasts at 0.01 nM.91
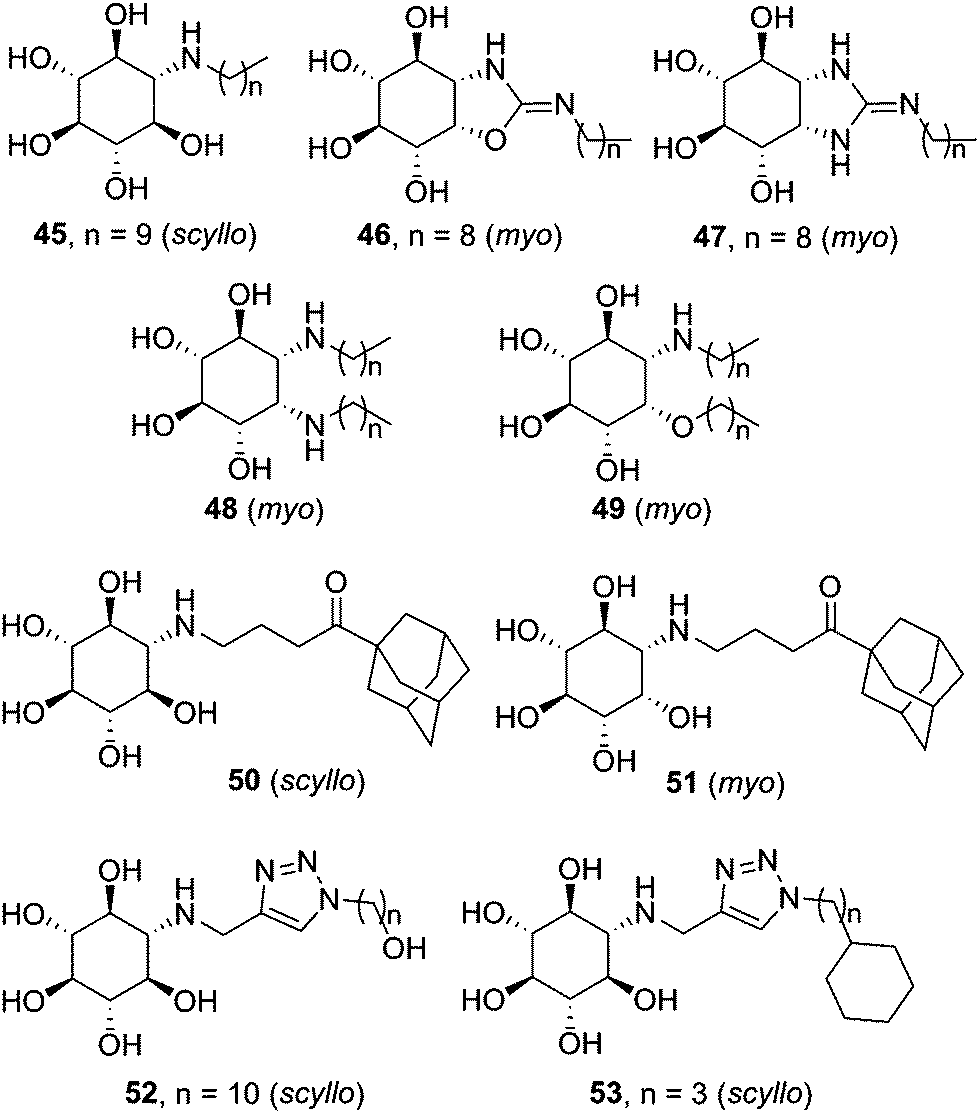 | ||
| Fig. 16 Structures of aminocyclitol derivatives developed as pharmacological chaperones for GD-associated mutant GCase variants. | ||
The data collection on aminocyclitols reveals the utmost importance of the configurational pattern and the nature of the exocyclic substituent in their inhibitory/chaperoning capabilities towards GCase. Amino-myo-inositols provided the best results in the bicyclic series, whereas both monocyclic scyllo- and myo-inositol derivatives showed promise as pharmacological chaperones for GD. Amphiphilicity is also a general requisite. In this sense, the incorporation of adamantanyl substituents, already found beneficial in iminosugar and sp2-iminosugar frameworks, represented an interesting option. Thus, compounds 50 (scyllo) and 51 (myo) induced outstanding 120% and 150% GCase activity increases, respectively, in homozygous L444P fibroblasts when used at 50 and 100 μM concentrations.92 Alternatively, the CuAAC click-type reaction can be used to generate molecular diversity at the N-alkyl moiety. The resulting 1,2,3-triazol adducts, e.g. amino-scyllo-inositols 52 and 53 (Fig. 16), were particularly efficient as chaperones in G202R/G202R GD fibroblasts, reaching up to 90% mutant GCase activity enhancements.93
Aminosugars
The observation that the presence of a nitrogen atom in the glycone moiety of glycomimetics is not a sine qua non prerequisite for strong GCase binding led Castillón, Ortiz Mellet and coworkers to propose the pyranose ring typical of monosaccharides as a scaffold for chaperone design.94 In principle, this approach should warrant glycone-driven specificity towards the target glycosidase while greatly simplifying the synthetic scheme. As a proof of concept, these authors developed derivatives with the cis-1,2-fused (gluco)pyranose-2-alkylsulfanyl-1,3-oxazoline (PSO) structure (54; Fig. 17) as conformationally locked N-glycoside analogues and demonstrated that their GCase inhibitory activity was drastically dependent on the nature of the exocyclic S-substituent. Installation of a hexadecyl chain with either a terminal hydroxy (PSO-HHD; 55) or a fluoro group (PSO-FHD; 56) provided the stronger GCase inhibitors within the series (IC50 values 12 and 3.9 μM, respectively), suggesting that PSO:GCase complexes might be stabilized by a long-range hydrogen bonding interaction involving this terminal group and an amino acid residue located at a maximum distance of 22 Å from the catalytic site (Fig. 17).95 PSO-HHD and PSO-FHD were able to enhance mutant GCase activity in N370S/N370S GD fibroblasts by 55% and 62% at 30 and 60 μM concentrations, respectively, doubling the maximum GCase activity enhancement of the non-glycomimetic chaperone Ambroxol®, currently in pilot trials in humans,96 in a parallel assay (33 μM at 10 μM).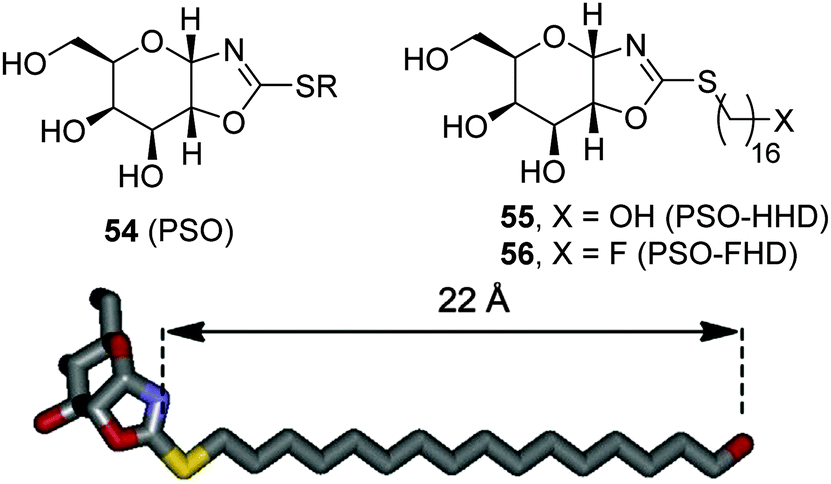 | ||
| Fig. 17 General structure of the PSO framework (54) and of the S-(ω-hydroxyhexadecyl) and S-(ω-fluorohexadecyl) derivatives 55 (PSO-HHD) and 56 (PSO-FHD), respectively (up), and 3D molecular model of PSO-HDD (carbons in grey, oxygens in red, nitrogen in light blue, sulfur in yellow; hydrogens have been omitted for the sake of clarity) with indication of the distance between the anomeric nitrogen atom and the terminal substituent in the chain (down). | ||
Following the same concept, Peregrina and coworkers97 proposed the aminopyrano[3,2-b]pyrrole (APP) framework 57 (Fig. 18) to build conformationally locked aminosugar C-glycosides onto which two different non-glycone substituents can be incorporated to optimize interactions with GCase. APP derivatives bearing one (O- or N-) or two palmitoyl chains behave as μM inhibitors of the enzyme, exhibiting one-order-of-magnitude decrease in the inhibition potency on going from pH 7 to pH 5, a favourable characteristic for pharmacological chaperone candidates. The N-palmitoyl derivative 58 was the most efficient APP within the series, promoting relative GCase activity increases of 1.3- and 1.5-fold in homozygous N370S and F213I GD fibroblasts, respectively, with no sign of toxicity. MD simulations of 58 in complex with the human enzyme supports the existence of a hydrogen bond involving the amide proton and the carboxylate group of the catalytic residue Glu235 that orients the palmitoyl chain towards a hydrophobic pocket in the enzyme (Fig. 18).
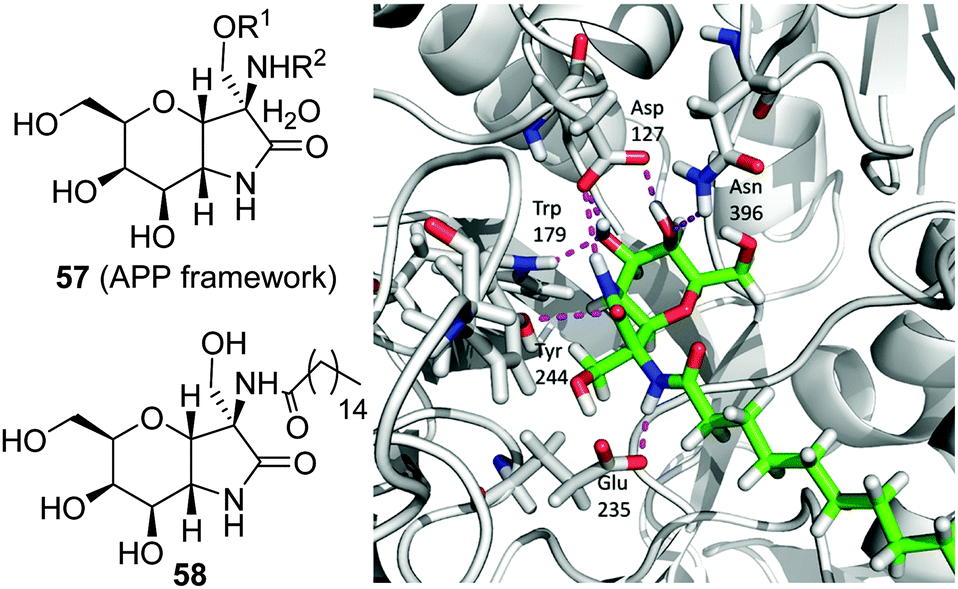 | ||
| Fig. 18 General structure of the APP framework (57) and of the N-palmitoyl derivative 58 (left); a representative frame obtained from MD simulations showing the hydrogen bonds between the glycone unit of compound 58 and amino acid residues in the human GCase binding site is also shown (right). | ||
Glycomimetic-based PCs for GM1-gangliosidosis
The potential of the galacto-configured iminosugar 1-deoxygalactonojirimycin (DGJ, 59) and its N-butyl derivative (NB-DGJ, 60; Fig. 19) as pharmacological chaperones for β-galactosidase mutants associated with GM1 was already explored by Suzuki and coworkers in 2001.98 Significant enzyme activity increases (2.1- to 6.1-fold) were observed in mouse fibroblasts expressing human GM1 β-Gal variants corresponding to the juvenile (R201C) or adult phenotypes (I51T, R201H or R457Q). The increase was detected also in human fibroblasts from patients with GM1-gangliosidosis with the I51T and R201C mutations (3- and 7-fold, respectively) after culture with 1 mM DGJ for 4 days. The rather high dose of iminosugar needed to have a relevant β-Gal activity enhancement, a limitation also encountered when using the monosaccharide D-galactose as the chaperone,99 and the lack of discrimination capabilities of DGJ between β- and α-galactosidases raise some concerns about unwanted side effects that jeopardize the progress of this approach into the clinics, however.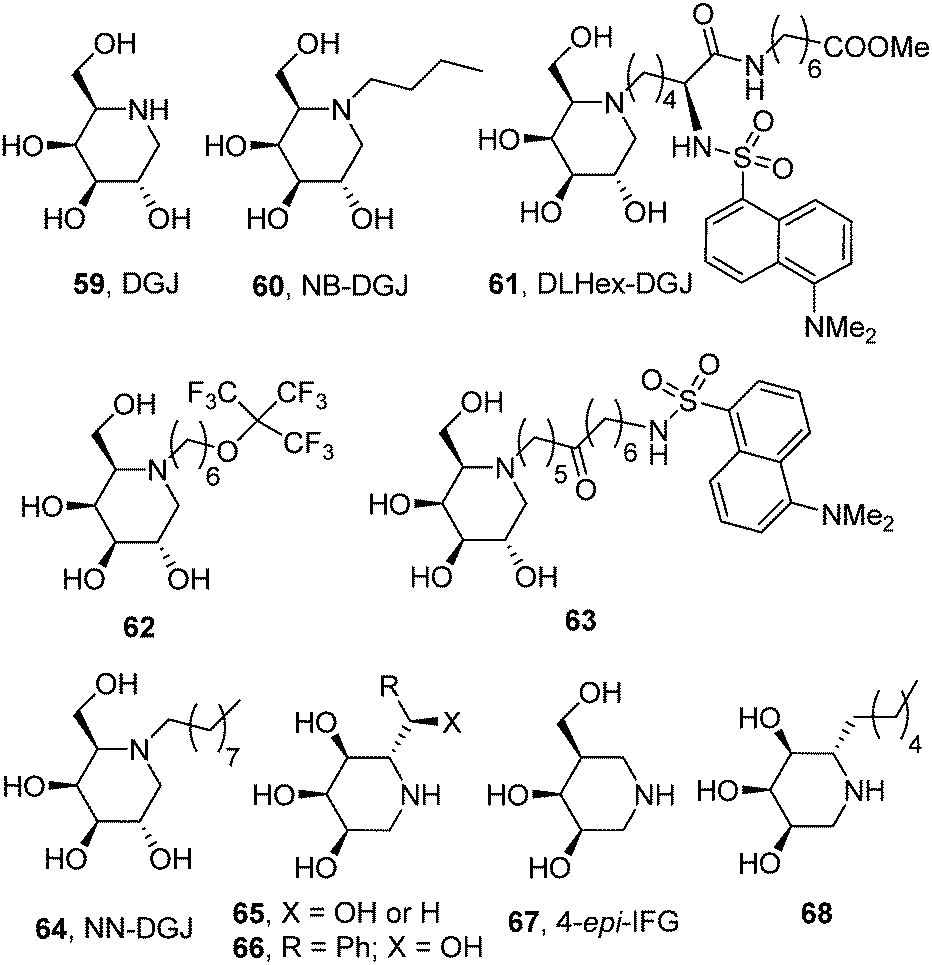 | ||
| Fig. 19 Structures of iminosugar-type PCs for GM1. | ||
Later on, Wroddning and coworkers investigated the effect of the nature of the N-alkyl substituent on the chaperoning properties of DGJ derivatives towards β-Gal mutants.100,101 This work led the authors to identify methyl 6-{[N2-(dansyl)-N6-(1,5-dideoxy-D-galactitol-1,5-diyl)-L-lysyl]amino} hexanoate (DLHex-DGJ, 61; Fig. 19) as a potent inhibitor of human lysosomal β-Gal (Ki 0.6 μM) significantly enhancing residual β-Gal activity (up to 18-fold) in homozygous R201C (juvenile GM1) and heterozygous R201H/R457X, R201H/H281Y and R201H/S149F fibroblasts (adult GM1).102 Although the maximum increase was achieved at a concentration of 500 μM, similar to that found for DGJ, in all single experiments the presence of DLHex-DGJ at 20 μM concentration was sufficient to yield more than 10% of the normal control mean activity, which was proposed as the critical level for normal substrate turnover. The minimal concentration of chaperone necessary to reach this threshold in homozygous R201C fibroblasts was further reduced to 5 μM for the fluorous derivative 62103,104 and to only 1 μM for the long-chain dansylated N-alkyl-DGJ derivative 63.105 Mahuran and coworkers reported a similar result for N-nonyl-DGJ (NN-DGJ, 64; Fig. 19).106 This amphiphilic iminosugar is a better inhibitor of β-Gal at neutral (IC50 6 nM) as compared to acidic pH (IC50 120 nM at pH 4.3) and produced a robust activity fold enhancement at 1.2 μM concentration in several GM1 fibroblasts derived from patients bearing the p.R201H/IVS14-2A>G (juvenile) and p.R201H/p.W509C (adult) mutations. The chaperoning capabilities of NN-DGJ iminosugar were also demonstrated in cells from a GM1 feline model.
In 2014 Siriwardena et al. proposed 1,5-dideoxy-1,5-iminoribitol (DIR) iminosugar C-glycosides with general structure 65 (R = alkyl, hydroxyalkyl or aryl; X = OH or H) as new chaperone candidates for GM1 (Fig. 19).107 The authors conceived a straightforward synthesis from D-ribose compatible with molecular diversity-oriented strategies and generated a small library of compounds from which selected members were assayed as β-Gal activity enhancers in fibroblasts from a patient with GM1-gangliosidosis heterozygous for the mutation p.R201H/IVS14-2A>G. The 2-hydroxy-2-phenylethyl derivative 66 exhibited the better chaperoning behavior within the series, eliciting enzyme activity increases over 6-fold when used at 394 μM concentration. Although no structural data are presented, the analogy of the DIR skeleton with the epimeric 1,5-dideoxy-1,5-iminoxilitol (DIX; Fig. 3) iminosugar core previously developed for Gaucher disease chaperone design is apparent, suggesting that the C-glycoside derivatives 65 probably bind to β-Gal in the 1-azasugar mode, that is, with the substituent at C-5 in a position equivalent to the N-substituent in DGJ derivatives. Recent results by Martin and coworkers further support this notion.108 These authors found that the galacto-configured glycomimetic 4-epi-isofagomine (67), a strong inhibitor of β-Gal (IC50 0.4 μM), was able to promote a 2.7-fold activity enhancement in GM1 fibroblasts harboring the R201C mutation when used at 10 μM concentration. Interestingly, the α-5-C-hexyl-DIR derivative 68 (Fig. 19), which was a much weaker inhibitor of the enzyme (IC50 100 μM), increased the mutant β-Gal activity by 2.1-fold at the same concentration in the same cell line, underlining the critical role of substituents at the C-5 position in controlling the activity of β-Gal chaperones in the 1-azasugar series.
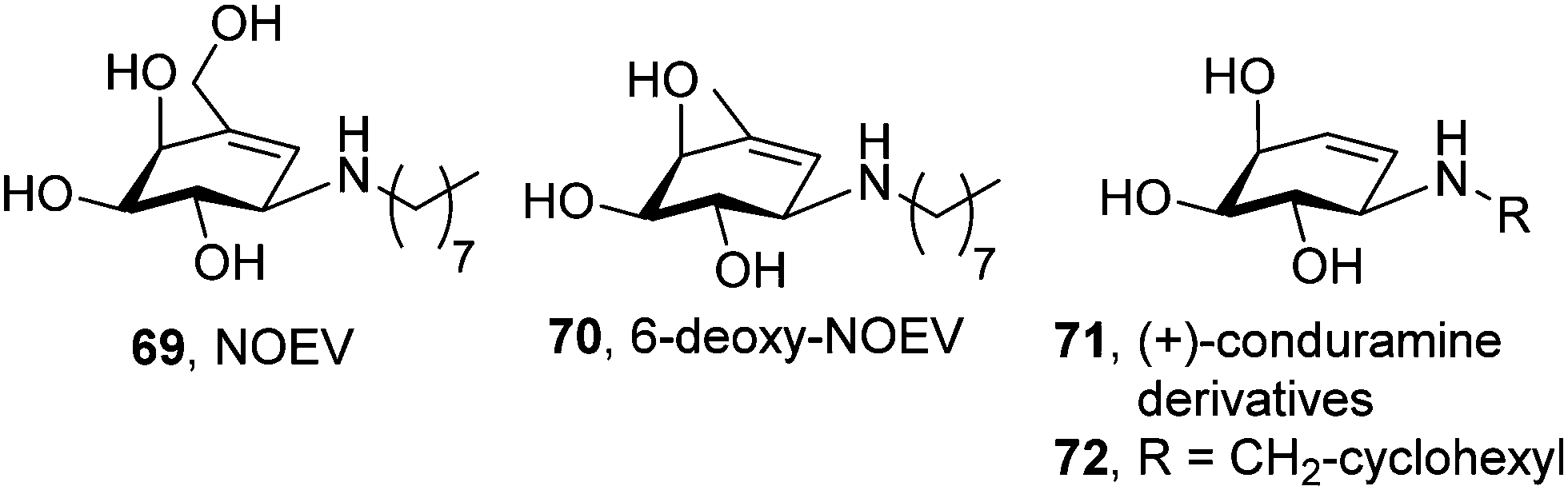
The synthesis in 2003 of N-octyl-epi-valienamine (NOEV, 69; Fig. 20) by Suzuki and coworkers and its characterization as a strong inhibitor of lysosomal β-galactosidase (IC50 0.2 μM) with very good chaperoning capabilities at low concentration (0.2 μM) represented a hallmark in the field of pharmacological chaperone therapy.109 NOEV, administered orally in water solution (1 mM) to mice expressing human R201C β-Gal ad libitum, produced an increase of enzyme activity and a parallel decrease in GM1-ganglioside in the cerebral cortex and brainstem. Accumulation of tropomyosin receptor kinase (Trk) receptors,110 p62 (a protein that regulates the formation of protein aggregates) and ubiquitinated proteins,111 which also contribute to GM1 pathogenesis, was also reduced after NOEV treatment. Most interestingly, NOEV treatment starting at the early stage of disease arrested the neurological progression of GM1 within a few months and resulted in a significantly prolonged survival time.112 Further results showed that the effect of NOEV can be synergistically enhanced by co-administration with the proteostasis regulators GM132 or celastrol.113
 | ||
| Fig. 20 Structures of NOEV, 6-deoxy-NOEV and (+)-conduramine derivatives developed as GM1 chaperones. | ||
The strong inhibitory character of NOEV may endanger its clinical effectiveness by counterbalancing the β-Gal activity enhancement in a dose-dependent manner. To cope with this problem, Kuno and coworkers114 modified the structure of the lead chaperone and prepared new analogues, including 6-deoxy-NOEV (70; Fig. 20) and a series of N-substituted (+)-conduramine derivatives (71), which combined much weaker β-Gal inhibition potencies with remarkable activity enhancements in fibroblasts from R201C GM1 patients. Compound 72, bearing an N-cyclohexylmethyl substituent, exhibited the best chaperone (8.5-fold activity increase) versus inhibitor (IC50 60 μM) ratio, although this result remains to be validated in vivo (Fig. 20).
Computational analysis for prediction of molecular interactions between the β-galactosidase protein and the chaperone compound NOEV were first conducted using a homology modeling method before the crystal structure of the protein was available.115 The data pointed to the critical contribution to complex stability of a hydrogen bonding between the amino group and the catalytic glutamic acid residue Glu188. Such interaction, which is similar to that further encountered for the β-oriented anomeric hydroxyl of its catalytic product D-galactose in the crystal structure of the corresponding complex with human β-Gal (Fig. 21A), is absent in DGJ (Fig. 21B), which explains its much weaker inhibition potency.116
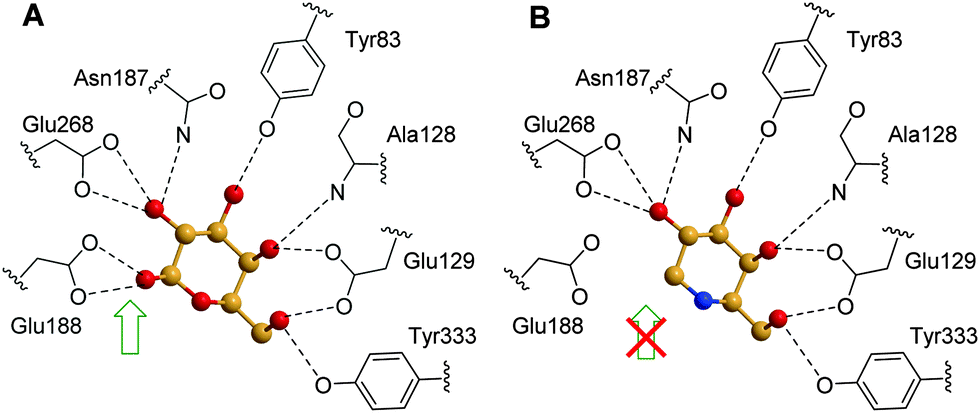 | ||
| Fig. 21 Schematic representation of the H-bonding network (dotted lines) of D-galactose (A) and DGJ (B) in the respective complexes with recombinant human β-Gal. The presence and absence of the key interaction with Glu188 is indicated with a green or red-crossed green arrow, respectively. | ||
Ortiz Mellet and coworkers117 conceived that the key hydrogen bonding interaction with the catalytic acid E188 could be restored in sp2-iminosugars having a bicyclic isothiourea-DGJ core, where the protonated exocyclic nitrogen would act as a donor. At the same time, a lipophilic substituent at this nitrogen can orient to the same hydrophobic pocket hosting the octyl chain in the NOEV:β-Gal complex. As a proof of concept, the 1-deoxygalactonojirimycin analogue 6S-NBI-DGJ (73, Fig. 22) exhibited strong and selective inhibition of β-Gal, with a ten-fold drop in affinity on going from pH 7 (IC50 3.1 μM) to pH 5 (IC50 32 μM). Gratifyingly, 6S-NBI-DGJ induced β-Gal activity enhancements comprised between 2- and 4-fold in fibroblasts from GM1 patients harboring the I51T/I51T, I51T/Y316C, I51T/R457Q, G190D/G190D, R201C/R201C, G438E/G438E or R457Q/R457Q mutations when used at 20–80 μM concentration.118 The only exception was encountered for cells with the R59H homozygous mutation. In a broader chaperone activity profiling on 88 types of human β-Gal mutants transiently expressed in COS-7 cells, up to 24 mutants were responsive to 6S-NBI-DGJ treatment, the other 64 being unresponsive. Noticeably, the mutations with positive responses included four common mutations, I51T, R201C, R208C, and R482H, suggesting that 6S-NBI-DGJ would be beneficial as a pharmacological chaperone for a large number of patients suffering from GM1-gangliosidosis. Therapeutic effects of 6S-NBI-DGJ in the brain of the R201C model mouse provided further evidence for its potential clinical impact. Thus, oral administration of 6S-NBI-DGJ at 5 and 10 mM significantly increased the mutant β-Gal activity in lysates from the cerebral cortex and brain stem of R201C mice and induced a remarkable reduction of lysosomal accumulation of GM1 ganglioside in the cortical sections of treated mice. The levels of autophagy-related proteins (LC3-II and p68) were also significantly reduced. These data strongly suggest that 6S-NBI-DGJ crossed the gastrointestinal and the blood–brain barriers and attained the central nervous system where it enhanced the activity of R201C mutant β-Gal and ameliorated the brain pathology.
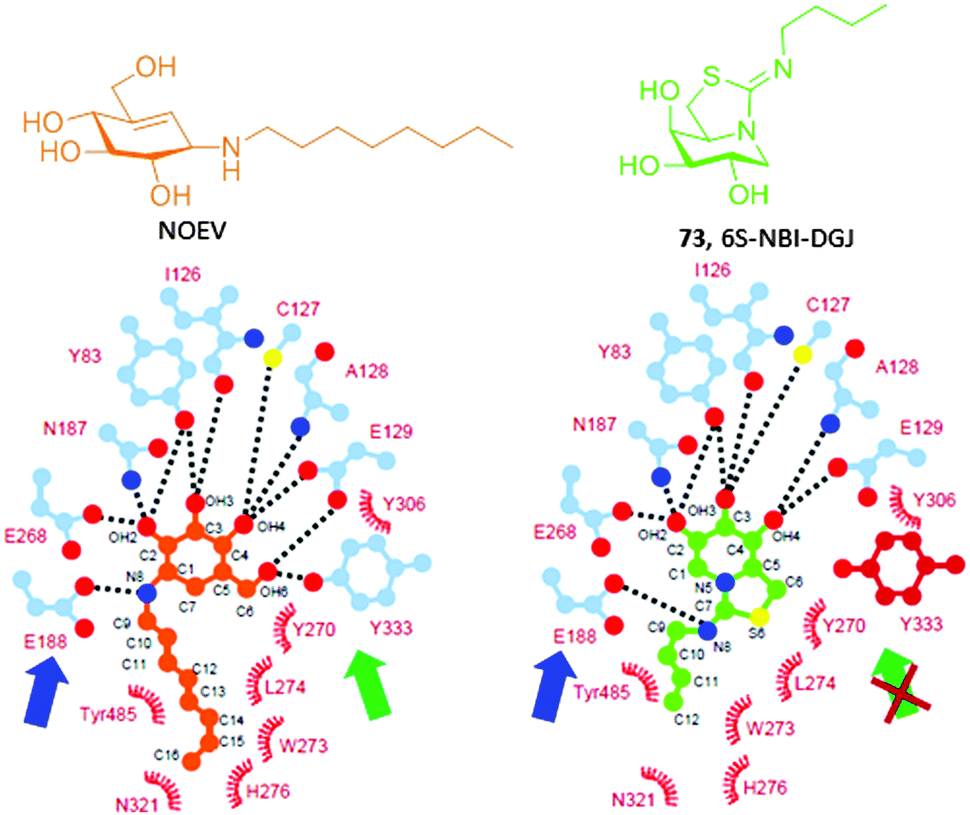 | ||
| Fig. 22 Schematic representation of the key H-bonding (dotted lines) and van der Waals interactions (arcs) in the complexes of NOEV (orange) and 6S-NBI-DGJ (green) in the respective complexes with recombinant human β-Gal. The catalytic glutamic acid residue E188, which is involved in the key interaction with the protonated amino or imino group in NOEV and 6S-NBI-DGJ is indicated with a blue arrow. The hydrogen bond implying the hydroxymethyl group in NOEV and Tyr133, which is absent for 6S-NBI-DGJ, is highlighted with a green or red-crossed green arrow, respectively. | ||
A comparative analysis of GM1-associated β-Gal mutations responsive to NOEV and to 6S-NBI-DGJ revealed significant differences in their chaperone activity profiles.119 For instance, NOEV was active as a chaperone for homozygous R201C but not I51T β-Gal whereas 6S-NBI-DGJ enhanced the enzyme activity of the two mutants in cell-based assays. The crystal structures of the corresponding complexes with β-Gal showed analogous hydrogen bonding networks at the catalytic site in both cases, including the hypothesized hydrogen bonding between the exocyclic nitrogen and E188, with the notable exception of the interaction involving the hydroxymethyl substituent and Tyr133, which is absent in the 6S-NBI-DGJ:β-Gal complex (Fig. 21). The authors suggested that this scenario confers a higher adaptability to the sp2-iminosugar chaperone that might be beneficial to optimally fit in and stabilize proper folding of some mutant forms of the enzyme.
Glycomimetic-based PCs for Fabry disease
In 1995, Okumiya et al. reported that galactose restores FD-associated mutant acid α-galactosidase (α-Gal A) activity.120 Subsequently, Fan et al. discovered that the iminosugar glycomimetic 1-deoxygalactonojirimycin (DGJ, Fig. 19), a potent inhibitor of α-Gal A (IC50 4 μM), restores the intracellular activity of mutant α-Gal A in cultured lymphoblasts from human hemizygous Fabry patients with the R301Q or Q279E mutations.121 DGJ is approximately 120000-fold more potent than galactose as a chaperone. The enzyme activity increased eight-fold or seven-fold (up to 48% or 45% of normal) after cultivation of R301Q or Q279E lymphoblasts, respectively, with DGJ at 20 μM for 4 days. However, DGJ at a concentration higher than 20 μM nullified the enhancement effect, highlighting the need of finely adjusting the dose to have an optimal chaperone versus inhibitory effect. The results were confirmed in COS-7 cells transfected with human R301Q α-Gal A cDNA and in fibroblasts of a transgenic mouse overexpressing human R301Q α-Gal A. When administered orally to the R301Q transgenic mice at 0.5 mM concentration, up to 18-fold α-Gal A activity increases were observed in the heart.
The above seminal work was followed by intense research in the chaperoning potential of DGJ against a broad range of disease-causing α-Gal A missense mutations in silico, in cellulo and in vivo.122–128 DGJ was α-Gal A specific, and did not affect misfolded mutant proteins in fibroblasts from other lysosomal storage disease patients at the concentrations effective for α-Gal A. Phase I clinical trials for DGJ (Amigal™) in healthy volunteers for safety and pharmacokinetics as well as several Phase II clinical trials with male and female Fabry patients who harboured a variety of missense mutations have already been accomplished.129,130 A Phase III clinical trial for Amigal™ in monotherapy is currently being conducted (data publication expected in 2016; http://www.amicustherapeutics.com). Such treatment (planned trade mark Galafold®) is expected to be highly effective for patients who have missense mutations that primarily lead to misfolding of the mutant protein (30% to 50% of Fabry patients). The aggregation-associated loss of function observed for the more severe α-Gal A mutants could be more efficiently overturned by combining pharmacological chaperone treatment with the suppression of mutant enzyme aggregation, e.g. by the combined use of proteostatic regulators.131 DGJ may also find efficacy in an adjunct therapy with manufactured α-Gal A used in enzyme replacement therapy (ERT) for patients whose residual enzyme activity cannot be increased by DGJ alone to a level that reverses disease development (Phase 3 clinical trial ongoing; http://www.amicustherapeutics.com). Thus, it was shown that coadministration of the chaperone and the enzyme slows the denaturation and loss in activity of α-Gal A ERT in vitro and increases uptake into cultured Fabry patient-derived cells. Most importantly, the combined therapy (DGJ orally administered, enzyme intravenously administered) increases the circulating levels of active recombinant α-Gal A used in enzyme replacement therapy in rodents and humans, results in higher tissue α-Gal A levels in mice and Fabry patients and leads to greater globotriaosylceramide (GL-3, Fig. 2) substrate reduction in disease-relevant tissues of Fabry mice compared to administration of α-Gal A in ERT alone.132,133 Similar benefits were reported for an intravenously administered DGJ-enzyme coformulation.134
DGJ has monopolized research on pharmacological chaperones for FD, with only a few alternative molecules on record. Sakuraba and coworkers conducted a comparative study of the chaperoning capabilities of DGJ and galactostatin bisulfite (74, GBS; Fig. 23) in COS-7 cells expressing the human M51I α-Gal A mutant.135 At 20 μM concentration both iminosugars increased enzyme activity by about 2.5-fold. On the other hand, the enzyme activity decreased when the concentration of the iminosugars was increased to 100 μM although the amount of protein remained the same, confirming that the iminosugars act as both pharmacological chaperones and inhibitors for the M51I mutant α-Gal A. SPR biosensor assaying further revealed that the rate dissociation constant (kd) value of the mutant α-Gal A:DGJ complex at pH 7.0 is almost the same as that at pH 5.0, strongly suggesting that there is still room for chaperone improvement, i.e., modification of the chemical structure for its quick dissociation from a mutant α-Gal A under acidic pH conditions.135
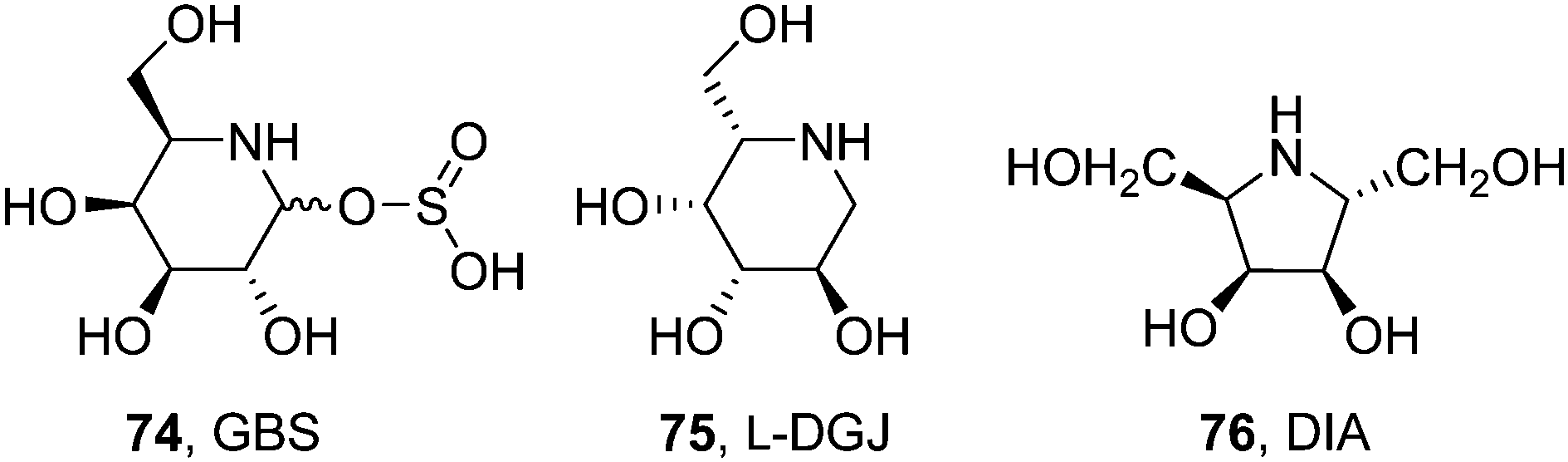 | ||
| Fig. 23 Structures of the piperidine iminosugars galactostatin bisulfate (GBS) and L-DGJ and the pyrrolidine iminosugar DIA showing FD-associated mutant α-Gal A chaperoning capabilities. | ||
Fleet, Kato and coworkers found that the enantiomer of DGJ, L-DGJ (75; Fig. 23), behaved as a noncompetitive inhibitor of α-Gal A (Ki 38.5 μM).136 Treatment of R301Q FD fibroblasts with 10 mM of L-DGJ increased intracellular α-Gal A activity by 10.8-fold, similar to that observed with 10 μM DGJ in a parallel assay. Most interestingly, each enantiomer did not counteract the chaperone effect of the other enantiomer, showing dose–response synergistic effects. In the pyrrolidine series, 2,5-dideoxy-2,5-imino-D-altritol (76, DIA; Fig. 23), a potent competitive inhibitor of human α-Gal A (Ki 0.5 μM) isolated from the roots of Adenophora triphylla, increased intracellular enzyme activity by 9.6-fold in Fabry R301Q lymphoblasts when used at 500 μM concentration.137
The crystal structure of DGJ bound to human α-Gal A revealed a hydrogen bonding interaction between the protonated endocyclic amino group and the carboxylate group of the catalytic aspartate nucleophile residue D170 that contributes very significantly to the stability of the complex (Fig. 24A).138 It was postulated that protonation of D170 in the lysosome might facilitate chaperone dissociation and substrate processing.139 Recent thermal shift assay experiments contradict this hypothesis, however, supporting that DGJ binds α-Gal A with similar affinities at neutral and at acidic pH.140 The authors conclude that DGJ is therefore not an optimal chaperone for Fabry disease, which is consistent with the prevalence of the inhibitory character at concentrations over 20 μM. In the case of a neutral ligand such as the monosaccharide galactose, the carboxylic acid catalytic residue D231, opposite to D170, stabilizes the corresponding complex by hydrogen bonding with the anomeric OH-1. Based on this structural evidence, Higaki, Ortiz Mellet and coworkers conceived that transformation of the basic amino group of DGJ into a neutral arylthiourea functionality might lead to a new family of α-Gal A inhibitors and chaperones, namely monocyclic DGJ-arylthioureas (DGJ-ArTs), better suited for drug optimization strategies (Fig. 24A).141 The strong hydrogen-bond donor capabilities of the NH thiourea proton, exacerbated by the electron withdrawing effect of the aromatic substituent, would reinforce the galactose-type complex stabilization mode.
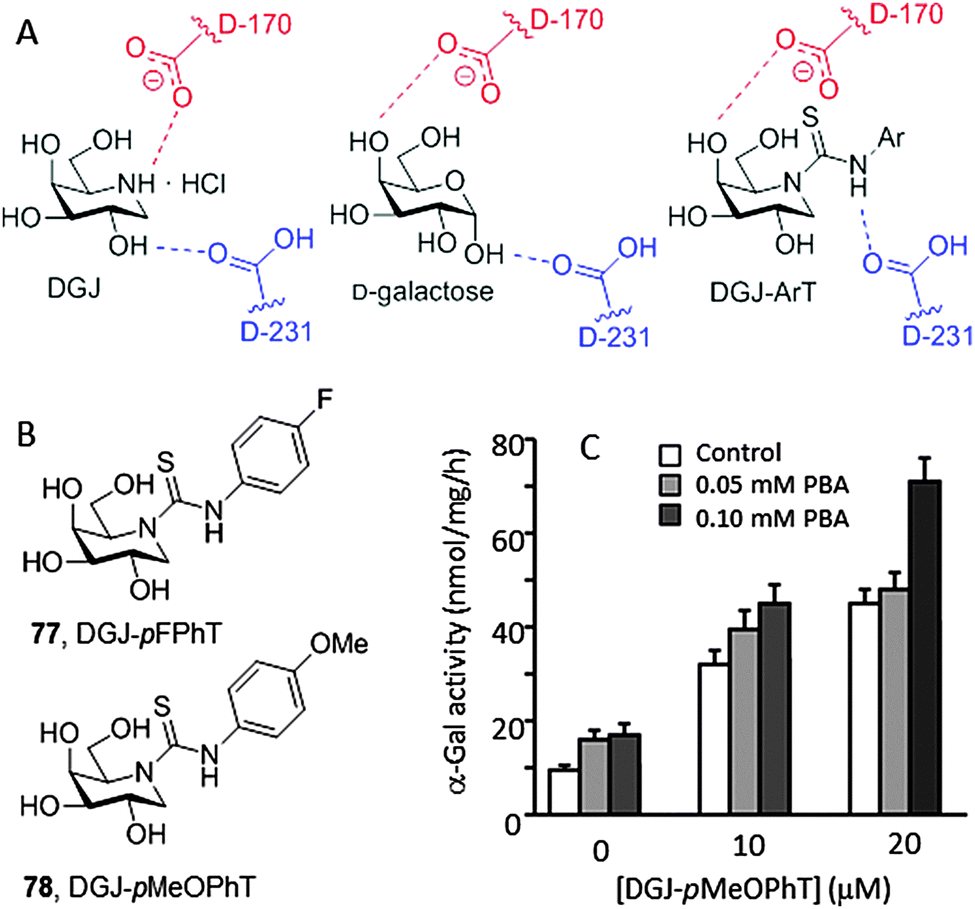 | ||
| Fig. 24 (A) Schematic representation of the main H-bonding interactions involving the catalytic acid aspartic acid residues in the complexes of DGJ, D-galactose and DGJ-arylthiourea (DGJ-ArT) derivatives with human α-Gal. (B) Structures of N′-p-fluorophenyl DGJ-ArT (77, DGJ-pFPhT) and N′-p-methoxyphenyl DGJ-ArT (78, DGJ-pMeOPhT). (C) Synergetic effect of the combined treatment with the chaperone DGJ-pMeOPhT and the proteostasis regulator 4-phenylbutiric acid (PBA) on SV-40-transformed Q279E Fabry disease fibroblasts. | ||
The crystal structure of the complex between α-Gal and the N′-p-fluorophenyl DGJ-ArT representative (77, DGJ-pFPhT) supported the above hypothesis. This compound and the N′-p-methoxyphenyl analogue (78, DGJ-pMeOPhT) behaved as very efficient chaperones, promoting up to 7-fold enzyme activity increases in Fabry disease fibroblasts expressing α-Gal A with the Q279E or R301Q mutations as compared with untreated cells when used at 30 μM, tripling the effect of DGJ at its optimal concentration of 20 μM. Immunofluorescence analyses revealed that treatment of Q279E fibroblasts with DGJ-ArTs significantly reduced the accumulation of globotriaosylceramide, indicating that the rescued enzyme is actively processing the substrate at the lysosome. To further investigate the chaperone effect of DGJ-ArTs on a range of mutant α-Gal A, a wild-type and 17 types of missense mutants were transiently transfected into COS-7 cells, with the activities of at least 15 out of the 17 mutants being significantly enhanced. Additionally, DGJ-ArT chaperones were efficient at correcting impairment of autophagy, a well-established hallmark of cellular pathology in lysosomal storage diseases. Q279E FD fibroblasts were first transformed with simian virus-40 (SV-40) to avoid inconveniences derived from senescence and then treated with the sp2-iminosugars. A significant reduction of the levels of the autophagy-related proteins LC3-II and p62 was observed. In addition, the DGJ-ArTs were found to act in a synergetic manner with the proteostasis regulator 4-phenylbutiric acid (4-PBA), confirming their potential in new Fabry disease therapeutic strategies (Fig. 24B).141
Conclusions and future outlook
In 2001, only six years since the first report on the chaperoning capabilities of galactose towards some FD-associated α-Gal A mutants,120 Desnik and coworkers published the first successful example of PC therapy in a patient: a 55-year-old man with the cardiac variant of Fabry disease who had residual α-Gal A activity as the result of a missense mutation (G328R) was given a galactose infusion (1 g per kilogram over a four-hour period every other day) and monitored for more than two years.142 At the beginning of the treatment, the subject had severe myocardial disease and was a candidate for cardiac transplantation. Galactose-infusion treatment resulted in increased α-Gal A activity (by 2- to 3-fold) in his circulating lymphocytes and endomyocardial cells, which translated into improved cardiac function. Cardiac transplantation was no longer required in this patient and he could return to full-time work as a bus driver. Deceivingly, after 20 years of intense research in the field, not a single drug is available for LSD treatment based on the PC concept. The files submitted to the FDA and EMA in 2016 by Amicus Therapeutics Inc. on Galafold® (DGJ) for Fabry patients harbouring responsive mutations might reverse this situation and pave the way to other glycomimetic PC candidates.Several hurdles can be identified that put at risk rapid translation of PC therapies into the clinics. Some are intrinsic to the neuronopathic character of most LSD conditions and the difficulties for drugs to cross the blood–brain barrier.143 Tuning PC amphiphilicity has been shown to allow reaching the central nervous system, but relatively high PC doses were needed to have a medically relevant result.118 Using targeted drug delivery devices, a strategy currently under investigation for ERT,144 may come in useful. Other limitations derive from the inability of the current glycomimetic PCs to comply with all the requirements for efficient functional protein rescuing, such as the need to program PCs to turn glucosidase binding from “on” at the ER to “off” at the final destination. Dissociation of the PC:glycosidase complex once in the lysosome is a prerequisite to allow substrate processing and turnover.145 The high initial substrate concentration in patient cells favors this exchange, but further equilibration can uncover the inhibitory character of active site-directed PCs, cancelling the chaperone benefit and thwarting a medically relevant result. Ortiz Mellet, García Fernández and coworkers have recently proposed an original chaperone prototype designed to undergo acid-promoted irreversible self-inactivation after mutant enzyme enhancement.146 The authors took advantage of the decisive contribution of hydrophobic non-glycone interactions in the stability of complexes between lysosomal glycosidases and sp2-iminosugar conjugates. Insertion of an acid-labile orthoester moiety in the structure let switching the nature of the chaperone from amphiphilic to hydrophilic in a biologically useful pH window. At pH 7, in the ER, the compound (e.g.79) exhibits strong affinity for the target mutant enzyme, being able to promote correct folding and facilitate trafficking. Once at the lysosome at pH below 5, the orthoester group is hydrolyzed, the hydrophobic segment is split off and the resulting hydrophilic derivative (e.g.80) is no longer a ligand of the enzyme and does not interfere with substrate processing. The concept was applied to monocyclic DNJ (Fig. 25) and DGJ sp2-iminosugar cores with high affinity towards human GCase and α-Gal A, respectively. Immunodetection proved that no inhibition effect occurred in fibroblasts of patients suffering from Gaucher or Fabry disease even at concentrations up to 200 μM, manifesting a pure chaperone behavior.146
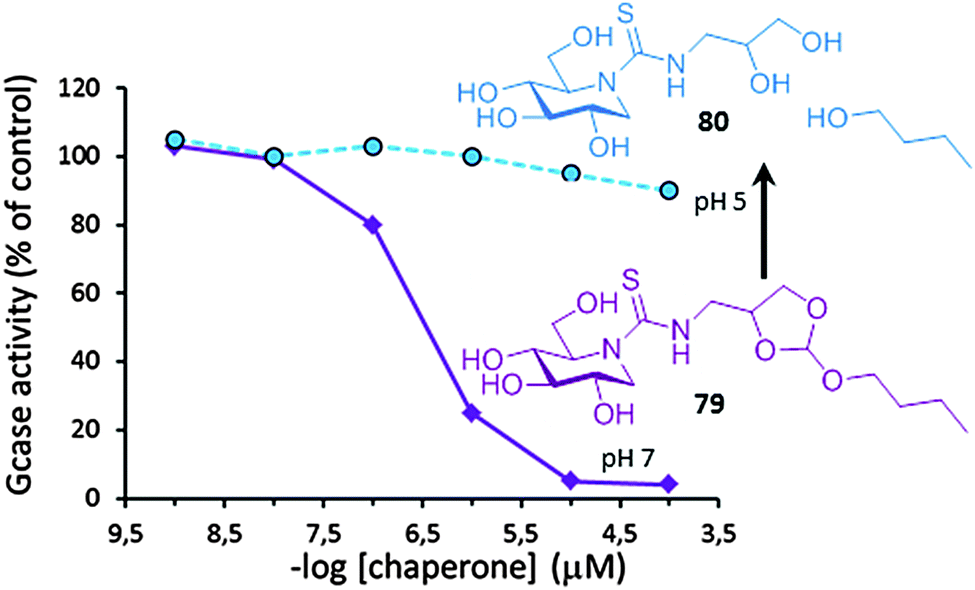 | ||
| Fig. 25 GCase inhibition plots for the orthoester-armed DGJ chaperone 79 at pH 7 (purple) and after incubation at pH 5 for 2 h (blue). HPLC monitoring confirmed that full hydrolysis into the inactive hydrophilic product 80 occurred upon neutral-to-acidic pH switch. | ||
The development of more reliable methods to assess chaperoning activity in situ, warranting that the measured enzyme increases translates into significant activity enhancements and substrate processing rates in the lysosomes, is contributing to accelerate PC optimization strategies.147 In this sense, there is an urgent need to establish protocols and initiatives for the reliable comparison of glycomimetics from different laboratories allowing confident selection of the best leads for preclinical studies. The contributions discussed in this Feature Article testify of the vitality of the field; although to avoid undue lengthening we have limited discussion to GD, GM1 and FD, glycomimetic-based PC candidates have also been developed for several other LSDs including Pompe,148 Krabbe,149 Tay-Sachs,150 Schindler/Kanzaki151 or Sanfilippo diseases.152 The increasing awareness that correction of lysosomal dysfunction might represent a therapeutic strategy for other neurodegenerative diseases such as Parkinson’s disease will likely stimulate research and open new avenues for glycomimetic PCs.153 The coming years will certainly witness significant progress in drug development from the concerted efforts of chemists, biochemists, structural biologists and biomedical researchers.
Acknowledgements
The Spanish Ministerio de Economía y Competitividad (MINECO, contract numbers SAF2013-44021-R and CTQ2015-64425-C2-1-R), the Junta de Andalucía (contract number FQM-1467), and the European Union Seventh Framework Programme (FP7-People-2012-CIG), grant agreement number 333594 (to E. M. S. F., Marie Curie Reintegration Grant) are acknowledged. Cofinancing from the European Regional Development Funds (FEDER and FSE) is also thanked.Notes and references
- F. M. Platt, B. Boland and A. C. van der Spoel, J. Cell Biol., 2012, 199, 723 CrossRef CAS PubMed.
- G. Parenti, G. Andria and A. Ballabio, Annu. Rev. Med., 2015, 66, 471 CrossRef CAS PubMed.
- F. M. Platt, Nature, 2014, 510, 68 CrossRef CAS PubMed.
- T. Wennekes, J. B. H. N. van den Berg, R. G. Boot, G. A. van der Marel, H. S. Overkleeft and J. M. F. G. Aerts, Angew. Chem., Int. Ed., 2009, 48, 8848 CrossRef CAS PubMed.
- D.-B. Oh, BMB Rep., 2015, 48, 438 CrossRef CAS PubMed.
- L. J. Scott, Drugs, 2015, 75, 1669 CrossRef CAS PubMed.
- F. M. Platt and M. Jeyakumar, Acta Paediatr., 2008, 97, 88 CrossRef PubMed.
- G. Parenti, G. Andria and K. J. Valenzano, Mol. Ther., 2015, 23, 1138 CrossRef CAS PubMed.
- J. A. Shayman and S. D. Larsen, J. Lipid Res., 2014, 55, 1215 CrossRef CAS PubMed.
- T. M. Gloster and D. J. Vocadlo, Nat. Chem. Biol., 2012, 8, 683 CrossRef CAS PubMed.
- C. Hetz and B. Mollereau, Nat. Rev. Neurosci., 2014, 15, 233 CrossRef CAS PubMed.
- R. E. Boyd, G. Lee, P. Rybczynski, E. R. Benjamin, R. Khanna, B. A. Wustman and K. J. Valenzano, J. Med. Chem., 2013, 56, 2705 CrossRef CAS PubMed.
- K. Mechler, W. K. Mountford, G. F. Hoffmann and M. Ries, Orphanet J. Rare Dis., 2015, 10, 46 CrossRef PubMed.
- B. H. Mele, V. Citro, G. Andreotti and M. V. Cubellis, Orphanet J. Rare Dis., 2015, 10, 55 CrossRef PubMed.
- P. Compain and A. Bodlenner, ChemBioChem, 2014, 15, 1239 CrossRef CAS PubMed.
- S. G. Gouin, Chem. – Eur. J., 2014, 20, 11616 CrossRef CAS PubMed.
- P. Compain, Synlett, 2014, 1215 CrossRef.
- B. Ernst and J. L. Magnani, Nat. Rev. Drug Discovery, 2009, 8, 661 CrossRef CAS PubMed.
- R. L. Lieberman, Enzyme Res., 2011, 2011, 973231 Search PubMed.
- A. Ciechanover and Y. T. Kwon, Exp. Mol. Med., 2015, 47, e147 CrossRef CAS PubMed.
- T. M. Cox, Biol.: Targets Ther., 2010, 4, 299 CrossRef CAS PubMed.
- E. B. Vitner, A. Vardi, T. M. Cox and A. H. Futerman, Expert Opin. Ther. Targets, 2015, 19, 321 CrossRef CAS PubMed.
- E. Sidransky, M. A. Nalls, J. O. Aasly, J. Aharon-Peretz, G. Annesi, E. R. Barbosa, A. Bar-Shira, D. Berg, J. Bras, A. Brice, C.-M. Chen, L. N. Clark, C. Condroyer, E. V. De Marco, A. Dürr, M. J. Eblan, S. Fahn, M. J. Farrer, H.-C. Fung, Z. Gan-Or, T. Gasser, R. Gershoni-Baruch, N. Giladi, A. Griffith, T. Gurevich, C. Januario, P. Kropp, A. E. Lang, G.-J. Lee-Chen, S. Lesage, K. Marder, I. F. Mata, A. Mirelman, J. Mitsui, I. Mizuta, G. Nicoletti, C. Oliveira, R. Ottman, A. Orr-Urtreger, L. V. Pereira, A. Quattrone, E. Rogaeva, A. Rolfs, H. Rosenbaum, R. Rozenberg, A. Samii, T. Samaddar, C. Schulte, M. Sharma, A. Singleton, M. Spitz, E.-K. Tan, N. Tayebi, T. Toda, A. R. Troiano, S. Tsuji, M. Wittstock, T. G. Wolfsberg, Y.-R. Wu, C. P. Zabetian, Y. Zhao and S. G. Ziegler, N. Engl. J. Med., 2009, 361, 1651 CrossRef CAS PubMed.
- N. Brunetti-Pierri and F. Scaglia, Mol. Genet. Metab., 2008, 94, 391 CrossRef CAS PubMed.
- Y. Suzuki, Brain Dev., 2011, 33, 719 CrossRef PubMed.
- M. Arends, C. E. M. Hollak and M. Biegstraaten, Orphanet J. Rare Dis., 2015, 10, 77 CrossRef PubMed.
- A. Jurecka and A. Tylki-Szymanska, Expert Opin. Orphan Drugs, 2015, 3, 293 CrossRef CAS.
- R. Lahiri, A. A. Ansari and Y. D. Vankar, Chem. Soc. Rev., 2013, 42, 5102 RSC.
- A. E. Stütz and T. M. Wrodnigg, Adv. Carbohydr. Chem. Biochem., 2011, 66, 187 CrossRef PubMed.
- G. Horne, F. X. Wilson, J. Tinsley, D. H. Williams and R. Storer, Drug Discovery Today, 2011, 16, 107 CrossRef CAS PubMed.
- Y. Sun, B. Liou, Y. H. Xu, B. Quinn, W. Zhang, R. Hamler, K. D. R. Setchell and G. A. Grabowski, J. Biol. Chem., 2012, 287, 4275 CrossRef CAS PubMed.
- A. Kato, S. Miyauchi, N. Kato, R. J. Nash, Y. Yoshimura, I. Nakagome, S. Hirono, S. Takahata and I. Adachi, Bioorg. Med. Chem., 2011, 19, 3558 CrossRef CAS PubMed.
- J. M. Benito, J. M. García Fernández and C. Ortiz Mellet, Expert Opin. Ther. Pat., 2011, 21, 885 CrossRef CAS PubMed.
- Y. Sun, H. Ran, B. Liou, B. Quinn, M. Zamzow, W. Zhang, J. Bielawski, K. Kitatani, K. D. R. Setchell, Y. A. Hannun and G. A. Grabowski, PLoS One, 2011, e19037 CAS.
- N. Dasgupta, Y.-H. Xu, R. Li, Y. Peng, M. K. Pandey, S. L. Tinch, B. Liou, V. Inskeep, W. Zhang, K. D. R. Setchell, M. Keddache, G. A. Grabowski and Y. Sun, Hum. Mol. Genet., 2015, 24, 7031 CAS.
- R. A. Steet, S. Chung, B. Wustman, A. Powe, H. Do and S. A. Kornfeld, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13813 CrossRef CAS PubMed.
- H.-H. Chang, N. Asano, S. Ishii, Y. Ichikawa and J.-Q. Fan, FEBS J., 2006, 273, 4082 CrossRef CAS PubMed.
- A. Kato, I. Nakagome, S. Nakagawa, Y. Koike, R. J. Nash, I. Adachi and S. Hirono, Bioorg. Med. Chem., 2014, 22, 2435 CrossRef CAS PubMed.
- T. S. Rasmusen and H. H. Jensen, Org. Biomol. Chem., 2010, 8, 433 Search PubMed.
- S. D. Orwig, Y. L. Tan, N. P. Grimster, Z. Yu, E. T. Powers, J. W. Kelly and R. L. Lieberman, Biochemistry, 2011, 50, 10647 CrossRef CAS PubMed.
- B. Brumshtein, H. M. Greenblatt, T. D. Butters, Y. Shaatiel, D. Aviezer, I. Silman, A. H. Futerman and J. L. Sussman, J. Biol. Chem., 2007, 282, 29052 CrossRef CAS PubMed.
- R. R. Wei, H. Hughes, S. Boucher, J. J. Bird, N. Guziewics, S. M. Van Patten, H. Qiu, C. Q. Pan and T. Edmunds, J. Biol. Chem., 2011, 286, 299 CrossRef CAS PubMed.
- M. N. Offman, M. Krol, I. Silman, J. L. Sussman and A. H. Futerman, J. Biol. Chem., 2010, 285, 42105 CrossRef CAS PubMed.
- R. J. Tamargo, A. Velayati, E. Goldin and E. Sidransky, Mol. Genet. Metab., 2012, 106, 257 CrossRef CAS PubMed.
- J. D. Diot, I. García Moreno, G. Twigg, C. Ortiz Mellet, K. Haupt, T. D. Butters, J. Kovensky and S. G. Gouin, J. Org. Chem., 2011, 76, 7757 CrossRef CAS PubMed.
- Click Chemistry in Glycoscience: New Developments and Strategies, ed. Z. J. Witczak and R. Bielski, Wiley-VCH, New Jersey, 2013, ISBN: 978-1-118-27533-7 Search PubMed.
- W.-C. Cheng, C.-Y. Weng, W.-Y. Yun, S.-Y. Chang, Y.-C. Lin, F.-J. Tsai, F.-Y. Huang and Y.-R. Chen, Bioorg. Med. Chem., 2013, 21, 5021 CrossRef CAS PubMed.
- G.-N. Wang, G. Reinkensmeier, S.-W. Zhang, J. Zhou, L.-R. Zhang, L.-H. Zhang, T. D. Butters and X.-S. Ye, J. Med. Chem., 2009, 52, 3146 CrossRef CAS PubMed.
- G.-N. Wang, G. Twigg, T. D. Butters, S. Zhang, L. Zhang, L.-H. Zhang and X.-S. Ye, Org. Biomol. Chem., 2012, 10, 2923 CAS.
- C. Parmeggiani, S. Catarzi, C. Matassini, G. D'Adamio, A. Morrone, A. Goti, P. Paoli and F. Cardona, ChemBioChem, 2015, 16, 2054 CrossRef CAS PubMed.
- X. Zhu, K. A. Sheth, S. Li, H.-H. Chang and J.-Q. Fan, Angew. Chem., Int. Ed., 2005, 44, 7450 CrossRef CAS PubMed.
- T. Hill, M. B. Tropak, D. Mahuran and S. G. Withers, ChemBioChem, 2011, 12, 2151 CrossRef CAS PubMed.
- P. Compain, O. R. Martin, C. Boucheron, G. Godin, L. Yu, K. Ikeda and N. Asano, ChemBioChem, 2006, 7, 1356 CrossRef CAS PubMed.
- A. Dondoni and A. Marra, Chem. Soc. Rev., 2012, 41, 573 RSC.
- E. D. Goddard-Borger, M. B. Tropak, S. Yonekawa, C. Tysoe, D. J. Mahuran and S. G. Withers, J. Med. Chem., 2012, 55, 2737 CrossRef CAS PubMed.
- E. Beutler, T. Gelbart and C. R. Scott, Blood Cells, Mol., Dis., 2005, 35, 355 CrossRef CAS PubMed.
- H. Dvir, M. Harel, A. A. McCarthy, L. Toker, I. Silman, A. H. Futerman and J. L. Sussman, EMBO Rep., 2003, 4, 704 CrossRef CAS PubMed.
- J. Serra-Vinardell, L. Díaz, J. Casas, D. Grinberg, L. Vilageliu, H. Michelakakis, I. Mavridou, J. M. F. G. Aerts, C. Decroocq, P. Compain and A. Delgado, ChemMedChem, 2014, 9, 1744 CAS.
- A. Kato, I. Nakagome, K. Sato, A. Yamamoto, I. Adachi, R. J. Nash, G. W. J. Fleet, Y. Natori, Y. Watanabe, T. Imahori, Y. Yoshimura, H. Takahatae and S. Hirono, Org. Biomol. Chem., 2016, 14, 1039 CAS.
- (a) M. Bergeron-Brlek, M. Meanwell and R. Britton, Nat. Commun., 2015, 6, 6903 CrossRef CAS PubMed; (b) S. Senthilkumar, S. S. Prasad, P. S. Kumar and S. Baskaran, Chem. Commun., 2014, 50, 1549 RSC; (c) A. Biela-Banas, E. Gallienne, S. Front and O. R. Martin, Tetrahedron Lett., 2014, 55, 838 CrossRef CAS; (d) M. Zoidi, B. Muller, A. Torvisco, C. Tysoe, M. Benazza, A. Siriwardena, S. G. Withers and T. Wrodnigg, Bioorg. Med. Chem. Lett., 2014, 24, 2777 CrossRef PubMed; (e) T. Wennekes, K. M. Bonger, K. Vogel, R. J. B. H. N. van den Berg, A. Strijland, W. E. Donker-Koopman, J. M. F. G. Aerts, G. A. van der Marel and H. S. Overkleeft, Eur. J. Org. Chem., 2012, 6420 CrossRef CAS; (f) P. Compain, V. Chagnault and O. R. Martin, Tetrahedron: Asymmetry, 2009, 20, 672 CrossRef CAS; (g) P. Merino, T. Tejero and I. Delso, Curr. Med. Chem., 2008, 15, 954 CrossRef CAS PubMed.
- T. M. Gloster and G. J. Davies, Org. Biomol. Chem., 2010, 21, 305 Search PubMed.
- M. I. García-Moreno, P. Díaz-Pérez, C. Ortiz Mellet and J. M. García Fernández, J. Org. Chem., 2003, 68, 8890 CrossRef PubMed.
- (a) E. M. Sánchez-Fernández, R. Rísquez-Cuadro, M. Aguilar-Moncayo, M. I. García-Moreno, C. Ortiz Mellet and J. M. García Fernández, Org. Lett., 2009, 11, 3306 CrossRef PubMed; (b) E. M. Sánchez-Fernández, R. Rísquez-Cuadro, M. Chasseraud, A. Ahidouch, C. Ortiz Mellet, H. Ouadid-Ahidouch and J. M. García Fernández, Chem. Commun., 2010, 46, 5328 RSC; (c) E. M. Sánchez-Fernández, R. Rísquez-Cuadro, C. Ortiz Mellet, J. M. García Fernández, P. M. Nieto-Mesa and J. Angulo-Álvarez, Chem. – Eur. J., 2012, 18, 8527 CrossRef PubMed; (d) G. Allan, H. Ouadid-Ahidouch, E. M. Sánchez-Fernández, R. Rísquez-Cuadro, J. M. García Fernández, C. Ortiz Mellet and A. Ahidouch, PLoS One, 2013, 8, e76411 CAS; (e) R. Rísquez-Cuadro, J. M. García Fernández, J.-F. Nierengarten and C. Ortiz Mellet, Chem. – Eur. J., 2013, 19, 16791 CrossRef PubMed; (f) E. M. Sánchez-Fernández, V. Gómez-Perez, R. García-Hernández, J. M. García Fernández, G. B. Plata, J. M. Padrón, C. Ortiz Mellet, S. Castanys-Cuello and F. Gamarro-Conde, RSC Adv., 2015, 5, 21812 RSC; (g) E. M. Sánchez-Fernández, E. Alvarez, C. Ortiz Mellet and J. M. García Fernandez, J. Org. Chem., 2014, 79, 11722 CrossRef PubMed; (h) E. M. Sánchez-Fernández, R. Gonalves-Pereira, R. Rísquez-Cuadro, G. B. Plata, J. M. Padrón, J. M. García Fernández and C. Ortiz Mellet, Carbohydr. Res., 2016 DOI:10.1016/j.carres.2016.01.006.
- V. M. Díaz Pérez, M. I. García-Moreno, C. Ortiz Mellet, J. Fuentes, J. M. García Fernández, J. C. Díaz Arribas and F. J. Cañada, J. Org. Chem., 2000, 65, 136 CrossRef.
- (a) J. L. Jiménez Blanco, V. M. Díaz Pérez, C. Ortiz Mellet, J. Fuentes, J. M. García Fernández, J. C. Díaz Arribas and F. J. Cañada, Chem. Commun., 1997, 1969 RSC; (b) J. M. García Fernández, C. Ortiz Mellet, J. M. Benito and J. Fuentes, Synlett, 1998, 316 CrossRef.
- (a) M. I. García-Moreno, J. M. Benito, C. Ortiz Mellet and J. M. García Fernández, J. Org. Chem., 2001, 66, 7604 CrossRef; (b) M. I. García-Moreno, C. Ortiz Mellet and J. M. García Fernández, Eur. J. Org. Chem., 2004, 1803 CrossRef.
- V. M. Díaz Pérez, M. I. García-Moreno, C. Ortiz Mellet and J. M. García Fernández, Synlett, 2003, 341 Search PubMed.
- M. I. García-Moreno, P. Díaz-Pérez, C. Ortiz Mellet and J. M. García Fernández, Chem. Commun., 2002, 848 RSC.
- (a) M. Aguilar-Moncayo, T. M. Gloster, J. P. Turkenburg, M. I. García-Moreno, C. Ortiz Mellet, G. J. Davies and J. M. García Fernández, Org. Biomol. Chem., 2009, 7, 2738 RSC; (b) M. Aguilar-Moncayo, M. I. García-Moreno, A. Trapero, M. Egido-Gabás, A. Llebaria, J. M. García Fernández and C. Ortiz Mellet, Org. Biomol. Chem., 2011, 9, 3698 RSC.
- (a) M. Aguilar, V. M. Díaz-Pérez, M. I. García-Moreno, C. Ortiz Mellet and J. M. García Fernández, J. Org. Chem., 2008, 73, 1995 CrossRef CAS PubMed; (b) R. Kooij, H. M. Branderhorst, S. Bonte, S. Wieclawska, N. I. Martin and R. J. Pieters, Med. Chem. Commun., 2013, 4, 387 RSC.
- M. Benltifa, M. I. García-Moreno, C. Ortiz Mellet, J. M. García Fernández and A. Wadouachi, Bioorg. Med. Chem. Lett., 2008, 18, 2805 CrossRef CAS PubMed.
- M. Aguilar-Moncayo, C. Ortiz Mellet, J. M. García Fernández and M. I. García-Moreno, J. Org. Chem., 2009, 74, 3595 CrossRef CAS PubMed.
- Z. Luan, K. Higaki, M. Aguilar-Moncayo, H. Ninomiya, K. Ohno, M. I. García-Moreno, C. Ortiz Mellet, J. M. García Fernández and Y. Suzuki, ChemBioChem, 2009, 10, 2780 CrossRef CAS PubMed.
- (a) B. Brumshtein, M. Aguilar-Moncayo, M. I. García-Moreno, C. Ortiz Mellet, J. M. García Fernández, I. Silman, Y. Shaaltiel, D. Aviezer, J. L. Sussman and A. H. Futerman, ChemBioChem, 2009, 10, 1480 CrossRef CAS PubMed; (b) B. Brumshtein, M. Aguilar-Moncayo, J. M. Benito, J. M. García Fernández, I. Silman, Y. Shaaltiel, D. Aviezer, J. L. Sussman, A. H. Futerman and C. Ortiz Mellet, Org. Biomol. Chem., 2011, 9, 4160 RSC.
- Z. Luan, K. Higaki, M. Aguilar-Moncayo, L. Li, H. Ninomiya, E. Nanba, K. Ohno, M. I. García-Moreno, C. Ortiz Mellet, J. M. García Fernández and Y. Suzuki, ChemBioChem, 2010, 11, 2453 CrossRef CAS PubMed.
- G. Tiscornia, E. Lorenzo Vivas, L. Matalonga, I. Berniakovich, M. Barragán Monasterio, C. E. Argaiz, L. Gort, F. González, C. Ortiz Mellet, J. M. García Fernández, A. Ribes, A. Veiga and J. C. Izpisua Belmonte, Hum. Mol. Genet., 2013, 22, 633 CrossRef CAS PubMed.
- M. Aguilar-Moncayo, M. I. García-Moreno, A. Trapero, M. Egido-Gabás, A. Llebaria, J. M. García Fernández and C. Ortiz Mellet, Org. Biomol. Chem., 2011, 9, 3698 CAS.
- J. Rodríguez-Lavado, M. de la Mata Fernández, J. L. Jiménez Blanco, M. I. García-Moreno, J. M. Benito, A. Díaz-Quintana, J. A. Sánchez Alcázar, K. Higaki, E. Nanba, K. Ohno, Y. Suzuki, C. Ortiz Mellet and J. M. García Fernández, Org. Biomol. Chem., 2014, 12, 2289 Search PubMed.
- B. Brumshtein, H. M. Greenblatt, T. D. Butters, Y. Shaaltiel, D. Aviezer, I. Silman, A. H. Futerman and J. L. Sussman, J. Biol. Chem., 2007, 282, 29052 CrossRef CAS PubMed.
- M. Aguilar, T. M. Gloster, M. I. García-Moreno, C. Ortiz Mellet, G. J. Davies, A. Llebaria, J. Casas, M. Egido-Gabás and J. M. García Fernández, ChemBioChem, 2008, 9, 2612 CrossRef CAS PubMed.
- P. Alfonso, V. Andreu, A. Pino-Ángeles, A. A. Moya-García, M. I. García-Moreno, J. C. Rodríguez-Rey, F. Sánchez-Jiménez, M. Pocoví, C. Ortiz Mellet, J. M. García Fernández and P. Giraldo, ChemBioChem, 2013, 14, 943 CrossRef CAS PubMed.
- (a) M. I. García-Moreno, C. Ortiz Mellet and J. M. García Fernández, Eur. J. Org. Chem., 2004, 1803 CrossRef; (b) M. I. García-Moreno, C. Ortiz Mellet and J. M. García Fernández, Tetrahedron, 2007, 63, 7879 CrossRef.
- M. De la Mata, D. Cotán, M. Oropesa-Ávila, J. Garrido-Maraver, M. D. Cordero, M. Villanueva Paz, A. Delgado Pavón, E. Alcocer-Gómez, I. de Lavera, P. Ybot-González, A. P. Zaderenko, C. Ortiz Mellet, J. M. García Fernández and J. A. Sánchez Alcázar, Sci. Rep., 2015, 5, 10903 CrossRef CAS PubMed.
- E. M. Govitrapong, P. Sharma, S. Muralikrishnan, D. Shavali, S. Pellett, L. Schafer, R. Albano and C. Eken, Biol. Signals Recept., 2001, 10, 224 CrossRef.
- L. Matalonga, A. Arias, M. J. Coll, J. Garcia-Villoria, L. Gort and A. Ribes, J. Inherited Metab. Dis., 2014, 37, 439 CrossRef CAS PubMed.
- H. Lin, H. Y. Sugimoto, Y. Ohsaki, H. Ninomiya, A. Oka, M. Taniguchi, H. Ida, Y. Eto, S. Ogawa, Y. Matsuzaki, M. Sawa, T. Inoue, K. Higaki, E. Nanba, K. Ohno and Y. Suzuki, Biochim. Biophys. Acta, 2004, 1689, 219 CrossRef CAS PubMed.
- Y. Sakakibara, S. Ogawa, K. Yugi, H. Jo and Y. Suzuki, J. Proteomics Bioinf., 2010, 3, 104 CrossRef.
- G. Sanchez-Olle, J. Duque, M. Egido-Gabas, J. Casas, M. Lluch, A. Chabas, D. Grinberg and L. Vilageliu, Blood Cells, Mol., Dis., 2009, 42, 159 CrossRef CAS PubMed.
- A. Trapero, I. Alfonso, T. D. Butters and A. Llebaria, J. Am. Chem. Soc., 2011, 133, 5474 CrossRef CAS PubMed.
- A. Trapero and A. Llebaria, ACS Med. Chem. Lett., 2011, 2, 614 CrossRef CAS PubMed.
- A. Trapero, P. Gonzalez-Bulnes, T. D. Butters and A. Llebaria, J. Med. Chem., 2012, 55, 4479 CrossRef CAS PubMed.
- A. Trapero, M. Egido-Gabás and A. Llebaria, Med. Chem. Commun., 2013, 4, 1584 RSC.
- J. Serra-Vinardell, L. Díaz, H. Gutiérrez-de-Terán, G. Sánchez-Ollé, J. Bujons, H. Michelakakais, I. Mavridou, J. M. F. G. Aerts, A. Delgado, D. Grinberg, L. Vilageliu and J. Casas, Int. J. Biochem. Cell Biol., 2014, 54, 245 CrossRef CAS PubMed.
- J. Castilla, R. Rísquez, D. Cruz, K. Higaki, E. Nanba, K. Ohno, Y. Suzuki, Y. Díaz, C. Ortiz Mellet, J. M. García Fernández and S. Castillón, J. Med. Chem., 2012, 55, 6857 CrossRef CAS PubMed.
- J. Castilla, R. Rísquez, K. Higaki, E. Nanba, K. Ohno, Y. Suzuki, Y. Díaz, C. Ortiz Mellet, J. M. García Fernández and S. Castillón, Eur. J. Med. Chem., 2015, 90, 258 CrossRef CAS PubMed.
- A. Zimran, G. Altarescu and D. Elstein, Blood Cells, Mol., Dis., 2013, 50, 134 CrossRef CAS PubMed.
- C. D. Navo, F. Corzana, E. M. Sánchez-Fernández, J. H. Busto, A. Avenoza, M. M. Zurbano, E. Nanba, K. Higaki, C. Ortiz Mellet, J. M. García Fernández and J. M. Peregrina, Org. Biomol. Chem., 2016, 14, 1473 CAS.
- L. Tominaga, Y. Ogawa, M. Taniguchi, K. Ohno, J. Matsuda, A. Oshima, Y. Suzuki and E. Nanba, Brain Dev., 2001, 23, 284 CrossRef CAS PubMed.
- A. Caciotti, M. A. Donati, A. d'Azzo, R. Salvioli, R. Guerrini, E. Zammarchi and A. Morrone, Eur. J. Paediatr. Neurol., 2009, 13, 160 CrossRef PubMed.
- G. Schitter, E. Scheucher, A. J. Steiner, A. E. Stütz, M. Thonhofer, C. A. Tarling, S. G. Withers, J. Wicki, K. Fantur, E. Paschke, D. J. Mahuran, B. A. Rigat, M. Tropak and T. M. Wrodnigg, Beilstein J. Org. Chem., 2010, 6, 21, DOI:10.3762/bjoc.6.21.
- R. F. G. Fröhlich, R. H. Furneaux, D. J. Mahuran, R. Sa, A. E. Stütz, M. B. Tropak, J. Wicki, S. G. Withers and T. M. Wrodnigg, Carbohydr. Res., 2011, 346, 1592 CrossRef PubMed.
- K. Fantur, D. Hofer, G. Schitter, A. J. Steiner, B. M. Pabst, T. M. Wrodnigg, A. E. Stütz and E. Paschke, Mol. Genet. Metab., 2010, 100, 262 CrossRef CAS PubMed.
- G. Schitter, A. J. Steiner, G. Pototschnig, E. Scheucher, M. Thonhofer, C. A. Tarling, S. G. Withers, K. Fantur, E. Paschke, D. J. Mahuran, B. A. Rigat, M. B. Tropak, C. Illaszewicz, R. Saf, A. E. Stutz and T. M. Wrodnigg, ChemBioChem, 2010, 11, 2026 CrossRef CAS PubMed.
- K. M. Fantur, T. M. Wrodnigg, A. E. Stütz, B. M. Pabst and E. Paschke, J. Inherited Metab. Dis., 2012, 35, 495 CrossRef CAS PubMed.
- R. F. G. Fröhlich, K. Fantur, R. H. Furneaux, E. Paschke, A. E. Stütz, J. Wicki, S. G. Withers and T. M. Wrodnigg, Bioorg. Med. Chem. Lett., 2011, 21, 6872 CrossRef PubMed.
- B. A. Rigat, M. B. Tropak, J. Buttner, E. Crushell, D. Benedict, J. W. Callahan, D. R. Martin and D. Mahuran, Mol. Genet. Metab., 2012, 107, 203 CrossRef CAS PubMed.
- A. Siriwardena, D. P. Sonawane, O. P. Bande, P. R. Markad, S. Yonekawa, M. B. Tropak, S. Ghosh, B. A. Chopade, D. J. Mahuran and D. D. Dhavale, J. Org. Chem., 2014, 79, 4398 CrossRef CAS PubMed.
- S. Front, E. Gallienne, J. Charollais-Thoenig, S. Demotz and O. R. Martin, ChemMedChem, 2016, 11, 133 CrossRef CAS PubMed.
- J. Matsuda, O. Suzuki, A. Oshima, Y. Yamamoto, A. Noguchi, Z. Takimoto, M. Itoh, Y. Matsuzaki, Y. Yasuda, S. Ogawa, Y. Sakata, E. Nanba, K. Katsumi, Y. Ogawa, L. Tominaga, K. Ohno, H. Iwasaki, H. Watanabe, R. O. Brady and Y. Suzuki, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 15912 CrossRef CAS PubMed.
- K. Takamura, H. Higaki, T. Ninomiya, J. Takai, J. Matsuda, M. Iida, K. Ohno, Y. Suzuki and E. Nanba, J. Neurochem., 2011, 118, 399 CrossRef PubMed.
- K. Higaki, L. Li, U. Bahrudin, S. Okuzawa, A. Takamuram, K. Yamamoto, K. Adachi, R. C. Paraguison, T. Takai, H. Ikehata, L. Tominaga, I. Hisatome, M. Iida, S. Ogawa, J. Matsuda, H. Ninomiya, Y. Sakakibara, K. Ohno, Y. Suzuki and E. Nanba, Hum. Mutat., 2011, 32, 843 CrossRef CAS PubMed.
- Y. Suzuki, S. Ichinomiya, M. Kurosawa, J. Matsuda, S. Ogawa, M. Iida, T. Kubo, M. Tabe, M. Itoh, K. Higaki, E. Nanba and K. Ohno, Mol. Genet. Metab., 2012, 106, 92 CrossRef CAS PubMed.
- L. Li, K. Higaki, H. Ninomiya, Z. Luan, M. Iida, S. Ogawa, Y. Suzuki, K. Ohno and E. Nanba, Mol. Genet. Metab., 2010, 101, 364 CrossRef CAS PubMed.
- S. Kuno, K. Higaki, A. Takahashi, E. Nanba and S. Ogawa, Med. Chem. Commun., 2015, 6, 306 RSC.
- Y. Suzuki, S. Ogawa and Y. Sakakibara, Perspect. Med. Chem., 2009, 3, 7 CAS.
- U. Ohto, K. Usui, T. Ochi, K. Yuki, K. Satow and T. Shimizu, J. Biol. Chem., 2012, 287, 1801 CrossRef CAS PubMed.
- M. Aguilar-Moncayo, T. Takai, K. Higaki, T. Mena-Barragán, Y. Hirano, K. Yura, L. Li, Y. Yu, H. Ninomiya, M. I. García-Moreno, S. Ishii, Y. Sakakibara, K. Ohno, E. Nanba, C. Ortiz Mellet, J. M. García Fernández and Y. Suzuki, Chem. Commun., 2012, 48, 6514 RSC.
- T. Takai, K. Higaki, M. Aguilar-Moncayo, T. Mena-Barragán, Y. Hirano, K. I. Yura, L. Yu, H. Ninomiya, M. I. García-Moreno, Y. Sakakibara, K. Ohno, E. Nanba, C. Ortiz Mellet, J. M. García Fernández and Y. Suzuki, Mol. Ther., 2013, 21, 526 CrossRef CAS PubMed.
- H. Suzuki, U. Ohto, K. Higaki, T. Mena-Barragan, M. Aguilar-Moncayo, C. Ortiz Mellet, E. Nanba, J. M. Garcia Fernandez, Y. Suzuki and T. Shimizu, J. Biol. Chem., 2014, 289, 14560 CrossRef CAS PubMed.
- T. Okumiya, S. Ishii, T. Takenaka, R. Kase, S. Kamei, H. Sakuraba and Y. Suzuki, Biochem. Biophys. Res. Commun., 1995, 214, 1219 CrossRef CAS PubMed.
- J.-Q. Fan, S. Ishii, N. Asano and Y. Suzuki, Nat. Med., 1999, 5, 112 CrossRef CAS PubMed.
- J.-Q. Fan and S. Ishii, FEBS J., 2007, 274, 4962 CrossRef CAS PubMed.
- J.-Y. Park, G.-H. Kim, S.-S. Kim, J. M. Ko, J.-J. Lee and H.-W. Yoo, Exp. Mol. Med., 2009, 41, 1 CrossRef CAS PubMed.
- E. R. Benjamin, J. J. Flanagan, A. Schilling, H. H. Chang, L. Agarwal, E. Katz, X. Wu, C. Pine, B. Wustman, R. J. Desnick, D. J. Lockhart and K. J. Valenzano, J. Inherited Metab. Dis., 2009, 32, 424 CrossRef CAS PubMed.
- R. Khanna, R. Soska, Y. Lun, J. Feng, M. Frascella, B. Young, N. Brignol, L. Pellegrino, S. A. Sitaraman, R. J. Desnick, E. R. Benjamin, D. J. Lockhart and K. J. Valenzano, Mol. Ther., 2010, 18, 23 CrossRef CAS PubMed.
- X. Wu, E. Katz, M. C. Della Valle, K. Mascioli, J. J. Flanagan, J. P. Castelli, R. Schiffmann, P. Boudes, D. J. Lockhart, K. J. Valenzano and E. R. Benjamin, Hum. Mutat., 2011, 32, 965 CrossRef CAS PubMed.
- B. Young-Gqamana, N. Brignol, H.-H. Chang, R. Khanna, R. Soska, M. Fuller, S. A. Sitaraman, D. P. Germain, R. Giugliani, D. A. Hughes, A. Mehta, K. Nicholls, P. Boudes, D. J. Lockhart, K. J. Valenzano and E. R. Benjamin, PLoS One, 2013, 8, e57631 CAS.
- E. R. Benjamin, J. J. Flanagan, A. Schilling, H. H. Chang, L. Agarwal, E. Katz, X. Wu, C. Pine, B. Wustman, R. J. Desnick, D. J. Lockhart and K. J. Valenzano, J. Inherited Metab. Dis., 2009, 32, 424 CAS.
- D. P. Germain, R. Giugliani, D. A. Hughes, A. Mehta, K. Nicholls, L. Barisoni, C. J. Jennette, A. Bragat, J. Castelli, S. Sitaraman, D. J. Lockhart and P. F. Boudes, Orphanet J. Rare Dis., 2012, 7, 91 CrossRef PubMed.
- R. Giugliani, S. Waldek, D. P. Germain, K. Nicholls, D. G. Bichet, J. K. Simosky, A. C. Bragat, J. P. Castelli, E. R. Benjamin and P. F. Boudes, Mol. Genet. Metab., 2013, 109, 86 CrossRef CAS PubMed.
- A. Siekierska, G. De Baets, J. Reumers, R. Gallardo, S. Rudyak, K. Broersen, J. Couceiro, J. Van Durme, J. Schymkowitz and F. Rousseau, J. Biol. Chem., 2012, 287, 28386 CrossRef CAS PubMed.
- E. R. Benjamin, R. Khanna, A. Schilling, J. J. Flanagan, L. J. Pellegrino, N. Brignol, Y. Lun, D. Guillen, B. E. Ranes, M. Frascella, R. Soska, J. Feng, L. Dungan, B. Young, D. J. Lockhart and K. J. Valenzano, Mol. Ther., 2012, 20, 717 CrossRef CAS PubMed.
- D. G. Warnock, D. G. Bichet, M. Holida, O. Goker-Alpan, K. Nicholls, M. Thomas, F. Eyskens, S. Shankar, M. Adera, S. Sitaraman, R. Khanna, J. J. Flanagan, B. A. Wustman, J. Barth, C. Barlow, K. J. Valenzano, D. J. Lockhart, P. Boudes and F. K. Johnson, PLoS One, 2015, 10, e0134341 Search PubMed.
- S. Xu, Y. Lun, N. Brignol, R. Hamler, A. Schilling, M. Frascella, S. Sullivan, R. E. Boyd, K. Chang, R. Soska, A. Garcia, J. Feng, H. Yasukawa, C. Shardlow, A. Churchill, A. Ketkar, N. Robertson, M. Miyamoto, K. Mihara, E. R. Benjamin, D. J. Lockhart, T. Hirato, S. Fowles, K. J. Valenzano and R. Khanna, Mol. Ther., 2015, 23, 1169 CrossRef CAS PubMed.
- T. Tsukimura, Y. Chiba, K. Ohno, S. Saito, Y. Tajima and H. Sakuraba, Mol. Genet. Metab., 2011, 103, 26 CrossRef CAS PubMed.
- S. F. Jenkinson, G. W. J. Fleet, R. J. Nash, Y. Koike, I. Adachi, A. Yoshihara, K. Morimoto, K. Izumori and A. Kato, Org. Lett., 2011, 13, 4064 CrossRef CAS PubMed.
- A. Kato, Y. Yamashita, S. Nakaqawa, Y. Koike, I. Adachi, J. Hollinshead, R. Nash, K. Ikeda and N. Asano, Bioorg. Med. Chem., 2010, 18, 3790 CrossRef CAS PubMed.
- R. L. Lieberman, J. A. D'Aquino, D. Ringe and G. A. Petsko, Biochemistry, 2009, 48, 4816 CrossRef CAS PubMed.
- A. I. Guce, N. E. Clark, J. J. Rogich and S. C. Garman, Chem. Biol., 2011, 18, 1521 CrossRef CAS PubMed.
- G. Andreotti, M. Monticelli and M. V. Cubellis, Drug Test. Analysis, 2015, 7, 831 CrossRef CAS PubMed.
- Y. Yu, T. Mena-Barragán, K. Higaki, J. Johnson, J. Drury, R. Lieberman, N. Nakasone, H. Ninomiya, T. Tsukimura, H. Sakuraba, Y. Suzuki, E. Nanba, C. Ortiz Mellet, J. M. García Fernández and K. Ohno, ACS Chem. Biol., 2014, 9, 1460 CrossRef CAS PubMed.
- A. F. Rustaci, C. Chimenti, R. Ricci, L. Natale, M. A. Russo, M. Pieroni, C. M. Eng and R. J. Desnick, N. Engl. J. Med., 2001, 345, 25 CrossRef PubMed.
- W. A. Banks, Nat. Rev. Drug Discovery, 2016 DOI:10.1038/nrd.2015.21.
- J. Hsu, T. Bhowmick, S. R. Burks, J. P. Y. Kao and S. Muro, J. Biomed. Nanotechnol., 2014, 10, 345 CrossRef PubMed.
- T. Mena-Barragán, M. I. García-Moreno, E. Nanba, K. Higaki, A. L. Concia, P. Clapés, J. M. García Fernández and C. Ortiz Mellet, Eur. J. Med. Chem., 2016 DOI:10.1016/j.ejmech.2015.08.038.
- T. Mena-Barragán, A. Narita, D. Matias, G. Tiscornia, E. Nanba, K. Ohno, Y. Suzuki, K. Higaki, J. M. Garcia Fernández and C. Ortiz Mellet, Angew. Chem., Int. Ed., 2015, 54, 11696 CrossRef PubMed.
- (a) A. K. Yadav, D. L. Shen, X. Shan, X. He, A. R. Kermode and D. J. Vocadlo, J. Am. Chem. Soc., 2015, 137, 1181 CrossRef CAS PubMed; (b) D. Herrera, M. Chao, W. W. Kallemeijn, A. R. A. Marques, M. Orre, R. Ottenhoff, C. van Roomen, E. Foppen, M. C. Renner, M. Moeton, M. van Eijk, R. G. Boot, W. Kamphuis, E. M. Hol, J. Aten, H. S. Overkleeft, A. Kalsbeek and J. M. F. G. Aerts, PLoS One, 2015, 10, e0138107 Search PubMed; (c) W. W. Kallemeijna, M. D. Witte, T. Wennekes and J. M. F. G. Aerts, Adv. Carbohydr. Chem. Biochem., 2014, 71, 297 CrossRef PubMed; (d) M. D. Witte, W. W. Kallemeijn, J. Aten, K.-Y. Li, A. Strijland, W. E. Donker-Koopman, A. M. C. H. van den Nieuwendijk, B. Bleijlevens, G. Kramer, B. I. Florea, B. Hooibrink, C. E. M. Hollak, R. Ottenhoff, R. G. Boot, G. A. van der Marel, H. Overkleeft and J. M. F. G. Aerts, Nat. Chem. Biol., 2010, 6, 907 CrossRef CAS PubMed.
- (a) G. Parenti, S. Fecarotta, G. la Marca, B. Rossi, S. Ascione, M. A. Donati, L. O. Morandi, S. Ravaglia, A. Pichiecchio, D. Ombrone, M. Sacchini, M. B. Pasanisi, P. De Filippi, C. Danesino, R. Della Casa, A. Romano, C. Mollica, M. Rosa, T. Agovino, E. Nusco, C. Porto and G. Andria, Mol. Ther., 2014, 22, 2002 CrossRef PubMed; (b) R. Khanna, A. C. Powe Jr., Y. Lun, R. A. Soska, J. Feng, R. Dhulipala, M. Frascella, A. Garcia, L. J. Pellegrino, S. Xu, N. Brignol, M. J. Toth, H. V. Do, D. J. Lockhart, B. A. Wustman and K. J. Valenzano, PLoS One, 2014, 9, e102092 Search PubMed; (c) R. Khanna, J. J. Flanagan, J. Feng, R. Soska, M. Frascella and L. J. Pellegrino, et al. , PLoS One, 2012, 7, e40776 CAS; (d) C. Bruckmann, H. Repo, E. Kuokkanen, H. Xhaard and P. Heikinheimo, ChemMedChem, 2012, 7, 1943 CrossRef CAS PubMed.
- (a) C. H. Hill, A. H. Viuff, S. J. Spratley, S. Salamone, S. H. Christensen, R. J. Read, N. W. Moriarty, H. H. Jensen and J. E. Deane, Chem. Sci., 2015, 6, 3075 RSC; (b) M. A. Hossain, K. Higaki, S. Saito, K. Ohno, H. Sakuraba, E. Nanba, Y. Suzuki, K. Ozono and N. Sakai, J. Hum. Genet., 2015, 60, 539 CrossRef CAS PubMed.
- (a) A. de la Fuente, R. Rísquez-Cuadro, X. Verdaguer, J. M. García Fernández, E. Nanba, K. Higaki, C. Ortiz Mellet and A. Riera, Eur. J. Med. Chem., 2016 DOI:10.1016/j.ejmech.2015.10.038; (b) J. S. S. Rountree, T. D. Butters, M. R. Wormald, S. D. Boomkamp, R. A. Dwek, N. Asano, K. Ikeda, E. L. Evinson, R. J. Nash and G. W. J. Fleet, ChemMedChem, 2009, 4, 378 CrossRef CAS PubMed.
- N. E. Clark, M. C. Metcalf, D. Best, G. W. J. Fleet and S. C. Garman, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17400 CrossRef CAS PubMed.
- M. Feldhammer, S. Durand and A. V. Pshezhetsky, PLoS One, 2009, 4, e7434 Search PubMed.
- R. E. Boyd and K. J. Valenzano, Bioorg. Med. Chem. Lett., 2014, 24, 3001 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |