
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New outcomes of Lewis base addition to diboranes(4): electronic effects override strong steric disincentives†
Nicole
Arnold
a,
Holger
Braunschweig
*a,
Alexander
Damme
a,
Rian D.
Dewhurst
a,
Leanne
Pentecost
b,
Krzysztof
Radacki
a,
Sascha
Stellwag-Konertz
a,
Torsten
Thiess
a,
Alexandra
Trumpp
a and
Alfredo
Vargas
b
aInstitut für Anorganische Chemie, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: h.braunschweig@uni-wuerzburg.de
bDepartment of Chemistry, School of Life Sciences, University of Sussex, Brighton BN1 9QJ, Sussex, UK
First published on 4th March 2016
Abstract
Two surprising new outcomes of the reaction of Lewis bases with dihalodiboranes(4) are presented, including sp2–sp3 diboranes in which the Lewis base unit is bound to a highly sterically congested boron atom, and a rearranged double base adduct. The results provide a fuller understanding of the reactivity of diboranes, a poorly-understood class of molecule of critical importance to synthetic organic chemistry.
After centuries of research, saturated acyclic chains of carbon atoms – alkanes – offer few surprises in contemporary organic chemistry. Likewise, other compounds with first-row main group homoatomic single bonds (N–N, O–O and F–F) are relatively well understood, albeit significantly more limited in number. In marked contrast, the remaining first-row main group element – boron – is distinctly reluctant to form such electron-precise, saturated homoatomic bonds. The chemistry of the simplest boron-based alkane analogues – diboranes(4) (R2BBR2) – is still in its infancy. Even setting aside the research that is providing a pipeline of borylated precursors for Suzuki–Miyaura cross-coupling reactions for the pharma, agrochemical and fine chemical industries,1 the field of diborane chemistry is witnessing strong growth spurred on by the frequent discovery of new capabilities of these compounds, such as substituent rearrangements, bridging interactions, bond activations and metal-free borylation reactions. A large amount of recent work and a number of reviews on the topics of B–B bond construction and the chemistry of sp2–sp3 diboranes highlight both the growing interest in this chemistry and its enormous potential.2
One of the most unpredictable classes of diboranes(4) are the dihalodiboranes(4), the substituents of which can rearrange both spontaneously and in the presence of bases.2,3 This process, involving the movement of substituents between sp2 atoms with empty p orbitals, is reminiscent of the Wagner–Meerwein rearrangement of alkyl groups in aliphatic carbocations.4 The first report of this behaviour came from the group of Berndt in 1991, wherein substitution of chlorides of symmetrical 1,2-diaryl-1,2-dichlorodiboranes(4) by fluoride proceeded with concomitant rearrangement to the unsymmetrical 1,1-diaryl-2,2-difluorodiboranes(4) (e.g.1, Fig. 1).5 The preference of the bulky aryl (mesityl, duryl) groups to both bind to the same boron atom was particularly surprising. More recently, we reported a number of similar rearrangements of 1,2-diaryl-1,2-dihalodiboranes(4) in the presence of Lewis bases, as well as isolation of base-stabilised diboranes(4) with halides bridging the boron atoms, assumed to be arrested rearrangement products.3 Reactions of bases with 1,2-dialkyl-1,2-dihalodiboranes(4) and 1,2-diamino-1,2-dihalodiboranes(4) uncovered further possibilities, such as simple (non-rearranged) adducts, C–H activation products and halide abstraction to form borylborenium cations. Together, these five outcomes represented a diverse range of structural outcomes from a seemingly simple reaction.
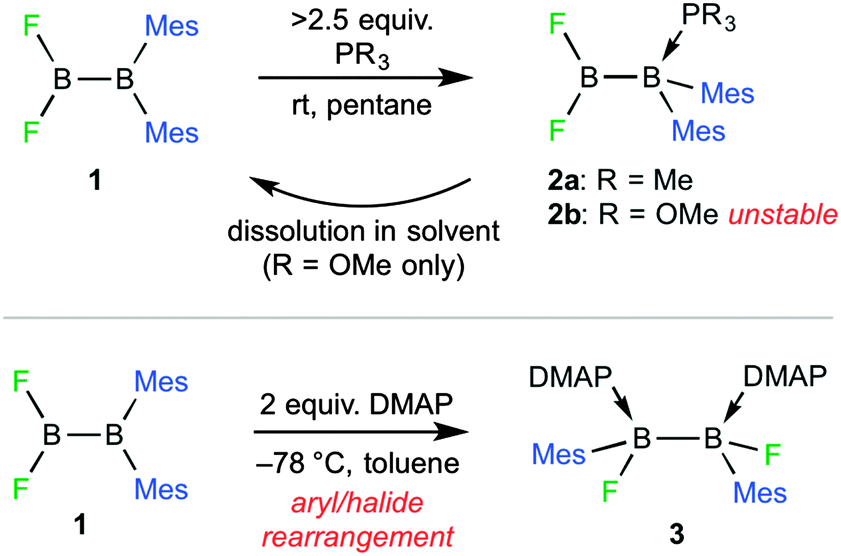 | ||
| Fig. 1 Synthesis of diborane adducts 2 and 3. DMAP = N,N-dimethylpyridine-4-amine. | ||
Herein, we report the surprising synthesis of adducts with Lewis bases bound instead to the sterically congested diaryl boron atom, as well as the formation of a (unsymmetrical-to-symmetrical) rearranged double base adduct. The preferences for base-ligation at the different boron atoms of 1,1-diaryl-2,2-dihalodiboranes are evaluated computationally in order to rationalise the observed results.
When the unsymmetrical diborane(4) 1,1-dimesityl-2,2-difluorodiborane(4) (1, Fig. 1)5 was treated with 5.8 equiv. of PMe3, 31P and 11B NMR spectra of the reaction mixture indicated conversion to a single boron-containing product, with both sp2 and sp3 boron centres (δB 35.9, −12.7; δP −16.4). Due to its high solubility in pentane, only a low yield of the isolated compound could be obtained (2a, Fig. 1). The downfield 11B NMR resonance of 2a at δB 35.9 is only moderately shifted from the corresponding resonance of precursor 1 (δB ∼ 29), suggesting that no rearrangement had taken place, while the upfield signal at δB −12.7 is shifted markedly from its original position (δB ∼82). That the downfield resonance of 1 shifts most upon base addition suggested that the base binds not to the BF2 unit but to the highly congested BMes2 unit. A similar reaction of 1 with 2.5 equiv. trimethylphosphite resulted in no observable conversion by 11B and 31P NMR spectroscopy. However, a low yield of colourless crystals was isolated from the reaction mixture that provided elemental analysis data consistent with a Lewis base mono-adduct and allowed determination of a solid-state structure (vide infra). Redissolving these crystals resulted in complete reversion to precursors 1 and P(OMe)3 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio, preventing the collection of NMR spectra.
:1 molar ratio, preventing the collection of NMR spectra.
The crystallographically-determined structures of 2a and 2b confirm the base addition at the diaryl-substituted boron centres (Fig. 2). Both structures suffer from disorder related to different orientations of the planar BF2 group, leading to unreliable B–F distances. The B–B distances of the compounds are identical within experimental uncertainty (2a: 1.704(3) Å; 2b: 1.711(4) Å), and, interestingly, are statistically identical to the B–B bond of the analogous, base-free diborane(4) B2F2Dur (Dur = 2,3,5,6-tetramethylphenyl; B–B 1.697(3) Å; see ESI,† for solid-state structure). The P–B distance of 2b (1.959(2) Å) is ca. 5% shorter than that of 2a (2.050(2) Å). Similarly small P–B distances in phosphite-boranes have been observed in the literature; for instance, the P–B bond in (MeO)3P→B(C6F5)3 (2.021(1) Å)6 is shorter than that of Me3P → B(C6F5)3 (2.061(4) Å),7 but only by ca. 2%. The sp3 boron atoms of 2a and 2b are strongly distorted from tetrahedral, with the sums of their B–B/C distances (2a ∑(∠B–X): 334.01°; 2b∑(∠B–X): 331.98°) being significantly higher than that expected for a perfect tetrahedron (328.5°). These angular sums indicate splayed BX3 units, while further distortion is indicated by the presence of one small and one large B–B–C angle in each structure (2a: B–B–C 99.2(1) and 120.8(1)°; 2b: B–B–C 95.5(2) and 124.0(2)°). This distortion is more pronounced in 2b, perhaps a consequence of its significantly shorter P–B bond, and is possibly a contributor to the observed instability of this compound. These parameters suggest a large amount of strain at the sp3 boron atom of both compounds, and that the connectivity observed is strongly disfavoured from a steric viewpoint.
 | ||
| Fig. 2 Crystallographically-derived structures of 2a, 2b, and 3, with thermal ellipsoids shown at the 50% probability level. Hydrogen atoms, ellipsoids of the mesityl groups, and solvent molecules (one molecule of CH2Cl2 for 3) are removed for clarity. Selected bond lengths (Å) and angles (°) for 2a: B1–B2 1.704(3), B1–F1 1.359(5), B1–F2 1.24(3), P1–B2 2.050(2); B1–B2-P1 103.9(1). For 2b: B1–B2 1.711(4), B1–F1 1.336(2), B1–F2 1.338(2), P1–B2 1.959(2); B1–B2–P1 102.3(2). For 3: B1–B2 1.794(4), B1–F1 1.444(3), B1–N1 1.649(3), B2–F2 1.434(3), B2–N2 1.652(3); B1–B2–N2 101.8(2), B1–B2–F2 110.2(2), B2–B1–N1 103.2(2), B2–B1–F1 109.1(2), torsion N1–B1–B2–N2 64.22, torsion F1–B1–B2–F2 152.45, torsion C1–B1–B2–C2 63.52. | ||
The apparent distortion in adducts 2 leads us to the conclusion that the binding site of the base is electronically controlled. While there is certainly no universal measure of Lewis acidity, BF3 is generally accepted to be a slightly stronger Lewis acid than BPh3,8 the electronics of which should not be vastly different from BMes3. It should be noted that the –BMes2 unit is used extensively in the synthesis of π-conjugated molecules and molecular materials, due to its combination of high π acidity and steric shielding that imparts kinetic protection from attack by nucleophiles,9 thus the observed base addition to this site is a surprising outcome.
In an attempt to prepare further examples of sp2–sp3 diboranes from 1, the diborane(4) was treated with 0.9 molar equivalents of N,N-dimethylpyridine-4-amine (DMAP). The reaction produces a number of different products, of which only the bis(base) adduct 3 could be isolated in low yield and fully characterised (Fig. 1). The yield of 3 could be increased to 27% by repeating the reaction with 2.0 equivalents of DMAP. The symmetrical nature of 3 is evident from its single 11B (δB 6.8) and 19F (δF −139.8) NMR resonances. The solid-state structure of 3 confirms the double coordination of DMAP units to the diborane(4) unit (Fig. 2). Looking down the B–B bond of 3, the six substituents are staggered, with the F groups approximately anti (torsion ca. 152°), the Mes groups syn (torsion ca. 63°), and the DMAP bases syn (torsion ca. 64°), respectively. The B–N distances of 3 (1.649(3), 1.652(3) Å) are very similar to those of bis(4-methylpyridine) adducts of diboranes(4) (ca. 1.64–1.66 Å).10 However, the B–B distance (1.794(4) Å) is significantly longer than those of the published compounds (ca. 1.71). It should be noted that doubly base-stabilised diboranes(4) are well known in the literature, including one example of a doubly base-stabilised diorganyldihalodiborane(4).11 However, complex 3 is distinguished from these examples by the fact that its synthesis involves the unusual unsymmetrical-to-symmetrical rearrangement.
Fig. 3 shows a chart of the outcomes of the reactions of dihalodiboranes with Lewis bases, with the products from this work shaded in yellow. The table highlights the remarkable diversity of the reaction when the relatively small differences between the precursors are considered. Changing the halide to fluoride in 1 clearly suppress the Lewis acidity of the attached boron atom such that the base chooses to bind at the diarylboryl site in compounds 2a,b or induces rearrangement in order to provide enough acidity to favour formation of dative N → B bonds in 3.
 | ||
| Fig. 3 Overall chart of the structurally diverse products of base addition to dihalodiboranes(4). Work presented herein is shaded in yellow, other results have been published previously.3 | ||
To better understand the isomeric selectivity of the diboranes, calculations were undertaken within the Kohn–Sham Density-Functional Theory (DFT). Fig. 4 shows the calculated relative total electronic energies of the different minimized isomers of (B2X2Mes2)(PMe3) (X = F, Cl, Br) adducts of type C and G as depicted in Fig. 3. The third minimised isomer for each halide case was found to be midway between the non-bridged type A (B–B–X angle >90°) and halide-bridged type B (with a B–B–X angle <90°). It can be immediately noticed that when X = F, type G is more stable than type C by ca. 13 kJ mol−1, although the hybrid isomer of type A/B is slightly more stable than G (by ca. 4 kJ mol−1). When X = Cl or Br, then type C is much more stable than G (by 45 or 64 kJ mol−1, respectively), while the hybrid type A/B is more stable than C by a further 12 or 4 kJ mol−1. These results corroborate the earlier-reported preference for X = Cl, Br, where the bridged adducts B are observed in the solid state. However, that the hybrid A/B structure is lower in energy in the X = F case suggests that the experimental absence of such a structure may be a kinetic obstacle, as to form such a compound one halide and one mesityl group of the unsymmetrical precursor 1 must switch places.
 | ||
| Fig. 4 Calculated relative total electronic energies (kJ mol−1) of molecules of types A/B, C and G with R = Mes, X = F, Cl, Br and L = PMe3. | ||
Computational techniques also provided plausible rationales for the unusual combination of high selectivity and highly variable structural outcomes observed in these diborane(4) Lewis adducts. Boron possesses unique abilities in terms of the ‘conveyance’ of charge flux,12 which are responsible for many of the outstanding properties of boron-containing compounds. Results from calculations suggest that the total electronic energy of diborane adducts is a function of the ‘charge-separation’ state of the B–B core. Indeed, Tables S1–S4 (ESI†) show that the sp2 boron atoms carry a positive partial charge, in contrast to the base-bound boron which in general carries a negative partial charge. It is clear that the boron partial charge depends on the nature of the neighbouring connected atoms or functional groups. The two fluorides of the BF2 unit strongly withdraw electron density from the boron, while in the BMes2 unit, the boron should support being slightly cationic as one Mes group can electronically stabilise it through resonance and induction, as evidenced by extensive σ and π interactions between B and the C1 atom of the one coplanar mesityl group. But because the boron atom interacting with the base is connected to another boron atom, the withdrawing effect presumably extends to the β boron atom. Hence the nature of the halide and the R group dictates at which acidic site the base binds in such a way as to have maximum BB charge difference,2d,13i.e., in a manner in which the acid–base interaction is strongest. This can be seen for instance by considering the B–L bond lengths. This reasoning is summed up in the following simple equation: Etot ∝ κ, where κ = qB(X) × qB(R) × q(b) × d(BB). In other words, the energy (and hence stability) depends on: the partial charge of both borons (reflecting the ease of charge movement conferred by the boron centres), the partial charge of the base (which takes into account its donating power) and lastly the boron–boron bond length (which takes into account any π(BB) donation, negative hyperconjugation between the BB substituents, etc.). Hybridisation does not seem to be very influential, as could be expected in the case of boron. The approach was tested and applied on model systems where R = Me, Ph. Setting R = Me, the κ values closely follow the trends in total energy; moreover, when X = F in the type C conformation, the base migrates to the diaryl side, indicating the inability of the methyl groups to stabilise a cationic B+ centre, in line with the scheme described above. In the case of R = Ph, the values are intermediate between those obtained when R = Mes and Me (data not shown). The validity of the model systems in terms of energetics and structure can be extended to the case where DMAP is used as base. When using the X = F, Cl models, the type C isomer is more stable than type G, as predicted using the aforementioned parameters.
In conclusion, the seemingly counterintuitive experimental observation of a Lewis base attacking the more sterically-hindered boron atom of an unsymmetrical diborane(4) can be ascribed to electronic effects. The results can be summarised as follows: (a) the system seeks to form the Lewis adduct where the donor–acceptor interaction is maximised (either side of the diborane), (b) this results in a ‘charge-separated’-like state within the BB unit, the cationic side being stabilised by σ and/or π electron donation, or by having strongly electronegative elements able to extend their σ-withdrawing effects to the boron in the β position.
These reactions further expand the remarkable number of outcomes of reactions between dihalodiboranes(4) and Lewis bases, and bring us closer to a more complete understanding of the chemistry of diboranes(4) – a class of molecule that is rapidly becoming a critical intermediate in synthetic organic chemistry.
Financial support for this work from the University of Sussex (A. V.) and the EPSRC (A. V.) is gratefully acknowledged.
Notes and references
- (a) Cross-coupling reactions of organoboranes: an easy way for C–C bonding (Nobel lecture), A. Suzuki, 2010, http://www.nobelprize.org/; (b) T. B. Marder and N. C. Norman, Top. Catal., 1998, 5, 63–73 CrossRef CAS; (c) Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials, ed. D. G. Hall, Wiley-VCH, Weinheim, Germany, 2006 Search PubMed; (d) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed; (e) J. F. Hartwig, Acc. Chem. Res., 2012, 45, 864–873 CrossRef CAS PubMed.
- (a) H. Braunschweig and R. D. Dewhurst, Angew. Chem., Int. Ed., 2013, 52, 3574–3583 CrossRef CAS PubMed; (b) H. Braunschweig, R. D. Dewhurst and S. Mozo, ChemCatChem, 2015, 7, 1630–1638 CrossRef CAS; (c) H. Gulyás, A. Bonet, C. Pubill-Ulldemolins, C. Solé, J. Cid and E. Fernández, Pure Appl. Chem., 2012, 84, 2219–2231 CrossRef; (d) J. Cid, H. Gulyás, J. J. Carbó and E. Fernández, Chem. Soc. Rev., 2012, 41, 3558–3570 RSC; (e) R. D. Dewhurst, E. C. Neeve, H. Braunschweig and T. B. Marder, Chem. Commun., 2015, 51, 9594–9607 RSC; (f) M. Gao, S. B. Thorpe and W. L. Santos, Org. Lett., 2009, 11, 3478–3481 CrossRef CAS PubMed; (g) A. Bonet, H. Gulyás and E. Fernández, Angew. Chem., Int. Ed., 2010, 49, 5130–5134 CrossRef CAS PubMed; (h) M. Gao, S. B. Thorpe, C. Kleeberg, C. Slebodnik, T. B. Marder and W. L. Santos, J. Org. Chem., 2011, 76, 3997–4007 CrossRef CAS PubMed; (i) S. B. Thorpe, X. Guo and W. L. Santos, Chem. Commun., 2011, 47, 424–426 RSC; (j) C. Kleeberg, A. G. Crawford, A. S. Batsanov, P. Hodgkinson, D. C. Apperley, M. S. Cheung, Z. Lin and T. B. Marder, J. Org. Chem., 2012, 77, 785–789 CrossRef CAS PubMed; (k) C. Borner and C. Kleeberg, Eur. J. Inorg. Chem., 2014, 2486–2489 CrossRef CAS; (l) S. Pietsch, E. C. Neeve, D. C. Apperley, R. Bertermann, F. Mo, D. Qiu, M. S. Cheung, L. Dang, J. Wang, U. Radius, Z. Lin, C. Kleeberg and T. B. Marder, Chem. – Eur. J., 2015, 21, 7082–7098 CrossRef CAS PubMed.
- (a) P. Bissinger, H. Braunschweig, A. Damme, R. D. Dewhurst, T. Kupfer, K. Radacki and K. Wagner, J. Am. Chem. Soc., 2011, 133, 19044–19047 CrossRef CAS PubMed; (b) H. Braunschweig, A. Damme, J. O. C. Jimenez-Halla, T. Kupfer and K. Radacki, Angew. Chem., Int. Ed., 2012, 51, 6267–6271 CrossRef CAS PubMed; (c) H. Braunschweig, A. Damme and T. Kupfer, Chem. Commun., 2013, 49, 2774–2776 RSC; (d) H. Braunschweig, A. Damme, R. D. Dewhurst, T. Kramer, T. Kupfer, K. Radacki, E. Siedler, A. Trumpp, K. Wagner and C. Werner, J. Am. Chem. Soc., 2013, 135, 8702–8707 CrossRef CAS PubMed.
- (a) J. Clayden, N. Greeves, S. Warren and P. Wothers, Organic Chemistry, Oxford University Press, Oxford, 2001 Search PubMed; (b) G. Wagner and W. Brickner, Ber. Dtsch. Chem. Ges., 1899, 32, 2302–2325 CrossRef CAS; (c) H. Meerwein, Liebigs Ann. Chem., 1914, 405, 129–175 CrossRef; (d) G. A. Olah, Acc. Chem. Res., 1976, 9, 41–52 CrossRef CAS.
- A. Höfner, B. Ziegler, R. Hunold, P. Willershausen, W. Massa and A. Berndt, Angew. Chem., Int. Ed., 1991, 30, 594–596 CrossRef.
- R. C. Neu, E. Y. Ouyang, S. J. Geier, X. Zhao, A. Ramos and D. W. Stephan, Dalton Trans., 2010, 39, 4285–4294 RSC.
- P. A. Chase, M. Parvez and W. E. Piers, Acta Crystallogr., 2006, E62, o5181–o5183 Search PubMed.
- (a) A. F. Holleman, E. Wiberg and N. Wiberg, Lehrbuch der Anorganischen Chemie, Walter de Gruyter, Berlin, New York, 2007, p. 1099 Search PubMed; (b) M. Méndez and A. Cedillo, Comput. Theor. Chem., 2013, 1011, 44–56 CrossRef; (c) I. B. Sivaev and V. I. Bregadze, Coord. Chem. Rev., 2014, 270–271, 75–88 CrossRef CAS.
- (a) Z. Yuan, J. C. Collings, N. J. Taylor, T. B. Marder, C. Jardin and J.-F. Halet, J. Solid State Chem., 2000, 154, 5–12 CrossRef CAS; (b) C. D. Entwistle and T. B. Marder, Angew. Chem., Int. Ed., 2002, 41, 2927–2931 CrossRef CAS; (c) C. D. Entwistle and T. B. Marder, Chem. Mater., 2004, 16, 4574–4585 CrossRef CAS; (d) Z. M. Hudson and S. Wang, Acc. Chem. Res., 2009, 42, 1584–1596 CrossRef CAS PubMed; (e) S. Yamaguchi and A. Wakamiya, Pure Appl. Chem., 2006, 78, 1413–1424 CAS; (f) F. Jäkle, Boron: Organoboranes, in Encyclopedia of Inorganic Chemistry, ed. R. B. King, Wiley, Chichester, U.K., 2nd edn, 2005 Search PubMed.
- (a) P. Nguyen, C. Dai, N. J. Taylor, W. P. Power, T. B. Marder, N. L. Pickett and N. C. Norman, Inorg. Chem., 1995, 34, 4290–4291 CrossRef CAS; (b) W. Clegg, C. Dai, F. J. Lawlor, T. B. Marder, P. Nguyen, N. C. Norman, N. L. Pickett, W. P. Power and A. J. Scott, J. Chem. Soc., Dalton Trans., 1997, 839–846 RSC.
- S. Pospiech, S. Brough, M. Bolte, H.-W. Lerner, H. F. Bettinger and M. Wagner, Chem. Commun., 2012, 48, 5886–5888 RSC.
- (a) H. Braunschweig, A. Damme, R. D. Dewhurst, S. Ghosh, T. Kramer, B. Pfaffinger, K. Radacki and A. Vargas, J. Am. Chem. Soc., 2013, 135, 1903–1911 CrossRef CAS PubMed; (b) H. Braunschweig, A. Damme, R. D. Dewhurst, T. Kramer, S. Östreicher, K. Radacki and A. Vargas, J. Am. Chem. Soc., 2013, 135, 2313–2320 CrossRef CAS PubMed.
- (a) A. Bonet, C. Pubill-Ulldemolins, C. Bo, H. Gulyás and E. Fernández, Angew. Chem., Int. Ed., 2011, 50, 7158–7161 CrossRef CAS PubMed; (b) J. Cid, J. J. Carbó and E. Fernández, Chem. – Eur. J., 2012, 18, 12794–12802 CrossRef CAS PubMed; (c) C. Pubill-Ulldemolins, A. Bonet, C. Bo, H. Gulyás and E. Fernández, Chem. – Eur. J., 2012, 18, 1121–1126 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and crystallographic details. CCDC 1452951–1452954. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc01493c |
| This journal is © The Royal Society of Chemistry 2016 |