
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign of a confined environment using protein cages and crystals for the development of biohybrid materials
Satoshi
Abe
,
Basudev
Maity
and
Takafumi
Ueno
*
Department of Biomolecular Engineering, Graduate School of Bioscience and Biotechonology, Tokyo Institute of Techonology, B-55, 4259 Nagatsuta-cho, Midori-ku, Yokohama 226-8501, Japan. E-mail: tueno@bio.titech.ac.jp
First published on 21st March 2016
Abstract
There is growing interest in the design of protein assemblies for use in materials science and bionanotechnology. Protein assemblies, such as cages and crystalline protein structures, provide confined chemical environments that allow immobilization of metal complexes, nanomaterials, and proteins by metal coordination, assembly/disassembly reactions, genetic manipulation and crystallization methods. Protein assembly composites can be used to prepare hybrid materials with catalytic, magnetic and optical properties for cellular applications due to their high stability, solubility and biocompatibility. In this feature article, we focus on the recent development of ferritin as the most promising molecular template protein cage and in vivo and in vitro engineering of protein crystals as solid protein materials with functional properties.
 Satoshi Abe | Satoshi Abe received his PhD from Nagoya University, Japan, in 2008. After working as a postdoctoral fellow at WPI-iCeMS at Kyoto University, he became an assistant professor at Tokyo Institute of Technology in 2012. His current research interests include the molecular design and engineering of protein crystals for the development of biohybrid materials. |
 Basudev Maity | Basudev Maity received his PhD from Indian Institute of Science (IISc), Bangalore, India, in 2012. Prior to joining as a JSPS postdoctoral research fellow at Tokyo Institute of Technology in 2013, he worked as a research associate at IISc, Bangalore. His current research interest is to design supramolecular protein assemblies for catalytic reactions. |
 Takafumi Ueno | Takafumi Ueno received his PhD from Osaka University, Japan, in 1998. He held a position as an assistant professor at the Institute for Molecular Science in Okazaki, Japan. From 2002 to 2008, he worked as an assistant professor at Nagoya University and then took a position as an associate professor at iCeMS at Kyoto University. In March 2012, he moved to the Tokyo Institute of Technology as a full professor. His current research interests include the molecular design of artificial metalloproteins and the exploitation of meso-scale materials using protein assemblies. |
1. Introduction
In recent years, strategies for the development of biomaterials using protein assemblies have attracted considerable attention in the fields of materials science and bio-nanotechnology due to their wide variety of applications.1–6 Highly ordered self-assembled proteins provide unique chemical environments for preparation of uniform nanomaterials and functionalization of metal complexes for applications in catalysis, imaging, and drug delivery, for example. Various protein assemblies, such as cages, tubes, and crystalline proteins, are utilized for such purposes.7 Although various synthetic templates, such as polymersomes, liposomes and dendrimers, are also known to have similar properties to nanomaterial scaffolds,8–11 the monodispersity, solubility and stability of protein assemblies under biological conditions are significantly distinct from those of synthetic materials. Among the various known classes of protein assemblies, we have particular interest in cages and crystalline protein assemblies for the following reasons; (i) the confined environments of proteins can accommodate various functional molecules, (ii) incorporation of the foreign substances into the interior space can be achieved by assembly/disassembly reactions, genetic manipulations, and protein crystallization methods, and (iii) these protein assemblies can be used in both in vivo and in vitro systems. In this feature article, we focus on recent significant development of ferritin cages as well as in vivo and in vitro protein crystal engineering.2. A protein cage for controlling cellular functions
Protein cages are formed from self-assembling proteins which provide a confined environment. Examples include virus capsid, heat shock protein (hsp), ferritin and DNA-binding protein from starved cells (dps) (Fig. 1a).12 Nature uses such confined protein compartments to protect interior molecules from the surrounding environment as observed in carboxysomes, in a manner similar to the function of organelles.13,14 The concept has been utilized to synthesize nanomaterials which can avoid self-aggregation.15,16 Similarly, small molecules or enzymes are incorporated into the cages.2,17 The advantages of using a protein cage include increased aqueous solubility, stability and biocompatibility of functional molecules when loaded into the cage.18 Virus capsids and ferritin are the two widely used protein cages for cellular applications. The size of the virus capsids or virus-like particles is larger (>20 nm) than that of the ferritin cage (12 nm) and often used for encapsulating larger substances like enzymes, polymeric materials, etc. for biological applications.17,19–23 For example, the interior cavity of the P22 capsid was used to incorporate Gd complexes embedded in the polymeric matrix for developing magnetic resonance imaging (MRI) contrast agents.24,25 Similarly, a CCMV capsid was used to incorporate a photosensitive self-immolative polymer which undergoes a head-to-tail depolymerization into its monomeric subunits when irradiated with light, resulting in the slow release of the molecular cargo.26 Chemotherapeutic drug taxol was functionalized in MS2 virus capsids which release the drug in MCF-7 cells.27 In contrast, the ferritin cage with a smaller cavity (8 nm) has specific metal binding sites and can be functionalized with a number of synthetic metal complexes or small organic molecules for biomedical applications. Importantly, the ferritin has specific cell targeting ability and internalizes through the transferrin receptor mediated endocytosis pathway.28 This is the important distinction of ferritin from other protein cages. Ferritin cages have potential applications in designing new materials, imaging, drug delivery, and catalysis.29 Several reviews have been published on the use of the protein cages such as ferritin cages and virus capsids as nanoreactors.3,18,30 Ferritin is a highly stable 24-mer self-assembled spherical protein cage.31 The outer diameter of the cage is 12 nm and the inner diameter is 8 nm (Fig. 1b). The natural function of ferritin is to store iron in a compact mineral form. The small 3-fold axis channels present in the ferritin cage provide the gateway for iron transport into the interior of the cage. It is known that a number of metal ions, metal complexes and organic molecules can be immobilized on the interior surface of apo-ferritin (apo-Fr) (Table 1). Due to their excellent biocompatibility, functionalized ferritin cages are used as carrier molecules for various biological applications (Table 1). The unique advantages of the ferritin cage are harnessed for the targeted delivery of small molecules and nanomaterials. We have focused our efforts on the ferritin cage which we believe is the most promising natural carrier protein cage. We discuss the recent advances in efforts to functionalize the ferritin cage for cellular applications. | ||
| Fig. 1 (a) Library of protein cages found in nature such as cowpea chlorotic mottle virus (CCMV; pdb ID: 1ZA7), heat shock protein (HSP; pdb ID: 1SHS), ferritin (pdb ID: 1DAT) and DNA binding protein from starved cells (DPS; pdb ID: 1QGH). (b) The intersection of the ferritin cage and a 3-fold symmetric pore. | ||
| Composites | Functions | Ref. |
|---|---|---|
| a nbd = norbornadiene. b Catecholamide = N-(2-(2,5-dioxo-1H-pyrrol-1-yl)ethyl)-2,3-dihydroxybenzamide. c RGD-4C = Cys-Asp-Cys-Arg-Gly-Asp-Cys-Phe-Cys. d NTTA = N,N,N1,N1-[40-(1-naphthyl)-2,20:60,20,0-terpyridine-6,60,0-diyl]bis(methylenenitrilo)tetrakis(acetic acid). e PTTA = N,N,N(1),N(1)-[[4′-phenyl-2,2′:6′,2′-terpyridine-6,6′-diyl]bis(methylenenitrilo)tetrakis(acetate)diyl]bis(methylenenitrilo)-tetrakis(acetate). f AF = Alexa Fluor. | ||
| PdNP | Olefin hydrogenation | 32 |
| Aerobic alcohol oxidation | 33 | |
| Au/Pd (core/shell) | Olefin hydrogenation | 34 |
| AuNP | Catalysis: nitro phenol reduction | 35 |
| Au nano cluster | Kidney specific targeting nanoprobe | 36 and 37 |
| Co doped ferrite NP | Theranostic agent | 38 |
| Fe3O4 | Targeting/imaging tumor tissue | 39 |
| Fe3O4/Cy5.5 | Imaging | 40 |
| CeO2 NP | Artificial redox enzyme activity | 41 |
| PtNP | Catalase and peroxidase activities | 42 and 43 |
| Anticancer drug | 44 | |
| PbS quantum dot | Antitumor activity | 45 |
| [Rh(nbd)Cl]2a | Polymerization of phenyl acetylene | 46 |
| [Pd(allyl)Cl]2 | Suzuki–Miyaura coupling | 47 |
| [Ru(CO)3Cl2]2 | CO releasing molecule | 48 |
| NF-κB activation | ||
| [Mn(CO)5Br] | CO releasing molecule | 49 |
| Light controlled NF-κB activation | ||
| Catecholamideb/FeCl3 | Siderophore mimic: enterobactin | 50 |
| Zn-phthalocyanine | Drug delivery: photodynamic therapy | 51 |
| Oxaliplatin | Drug delivery | 52 |
| K2PtCl4 | Drug delivery | 53 |
| Carboplatin | ||
| Cisplatin | ||
| Gd chelate complex | MRI contrast agent | 54–56 |
| Curcumim/Gd-chelate | Theranostic agent | 56 and 57 |
| Mn2+ | MRI sensor of melanin formation in melanoma cells | 58 |
| T 2 contrast agent | 59 | |
| RGD-4C,c Cy5.5 and 64Cu | Multimodal imaging | 60 |
| NTTAd-Eu3+ | Bioprobe for time-gated luminescence bioimaging | 61 |
| PTTAe-Tb3+ | Nitric oxide probe | 62 |
| Doxorubicin | Drug delivery | 63–65 |
| Methylene blue | Drug delivery: photodynamic therapy | 66 |
| Near-infrared dye IR820 | Multimodal imaging and photothermal therapy | 67 |
| AF350/AF430f | Fluorescence resonance energy transfer | 68 |
2.1 Nanomaterials in the ferritin cage
The uniform size of the ferritin cage is utilized to prepare monodisperse nanoparticles which have several applications in catalysis and imaging, among others. For example, uniform Pd nanoparticles have been prepared in a ferritin cage and used for catalytic olefin hydrogenation and an aerobic alcohol oxidation reaction.32–34 Biocompatible nanomaterials can be prepared using the ferritin template in one step. In contrast, conventional methods require additional capping or targeting ligands. The ferritin-nanoconjugates are easily taken up by cells expressing transferrin receptors and have been demonstrated to reach their targets. This is significant because targeting often represents a problem for most types of nanoparticles. Considering such advantages, several ferritin-nanocomposites have been developed for various cellular applications such as enzyme mimetics and bioimaging, among others.36,41,42,69SOD mimicking nanoceria (CeO2) particles have been incorporated into the ferritin cage via a dissociation–reconstruction route (Fig. 2a).41 The protein scaffold provides biocompatibility and higher cellular uptake via a receptor mediated pathway. The cage also manipulates the electron localization at the surface of nanoparticles. The ROS (reactive oxygen species) scavenging activity of apo-Fr encapsulated CeO2 is 3.5 times higher than that of the natural SOD enzyme. The activity was tested in HepG2 cells using DCFH-DA (2,7-dichlorofluorescein diacetate) dye which is used to measure the ROS level. The effective quenching of the green fluorescence of DCFH-DA suggests that the system has excellent ROS scavenging activity in living cells. The iron oxide core was prepared in the apo-Fr cage to develop ferritin containing magnetic particles (magnetoferritin) which has applications in targeting and visualizing tumor tissues.39 Since the iron core is formed inside the ferritin cage, the nanoparticles in magnetoferritin are easily taken up by overexpressed transferrin receptor 1 (TfR1) of tumor cells.
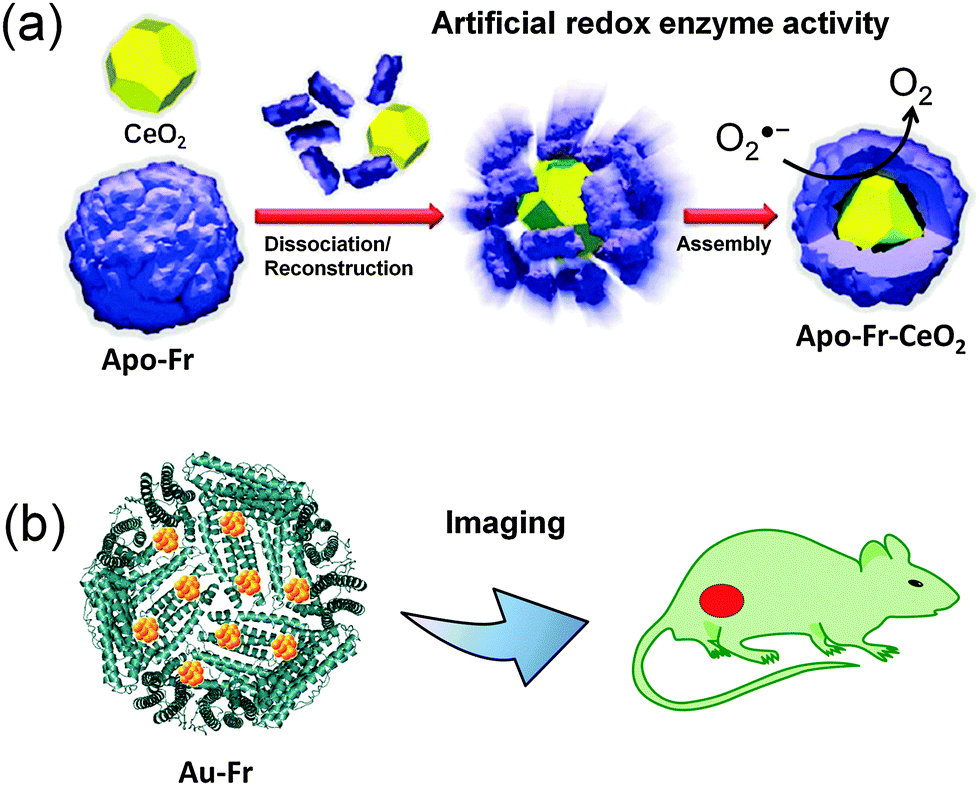 | ||
| Fig. 2 Applications of nanomaterials prepared in the ferritin cage. (a) The SOD mimicking apo-Fr-CeO2: preparation and functions. Reproduced from ref. 41 with permission from RSC. (b) Use of ferritin templated gold nanoclusters for bioimaging. | ||
The composite catalyzes the oxidation of peroxidase substrates such as 3,3,5,5-tetramethylbenzidine (TMB) or di-aza-aminobenzene (DAB) in the presence of hydrogen peroxide to produce colored products which are utilized for visualizing the tumor tissues. The formation of hydroxyl radicals on the surface of the iron core is thought to be responsible for the catalytic oxidation reaction. Magnetoferritin can be used as an efficient diagnostic tool for cancer cell detection in a number of clinical samples. In a recent report, it was demonstrated that iron oxide can be loaded into the ferritin cage and functionalized with near-infrared emitting dye (Cy5.5).40 The hybrid nanocomposite exhibits extremely high transversal relaxivity ((r2) up to 224 mM−1 s−1) and can cross the endothelium, epithelium and blood brain barrier layers. The composite can specifically target tumors overexpressing TfR1. Thus the ferritin based nanoprobe is used for targeted and ultra-sensitive imaging. There are several other reports describing the incorporation of Pt nanoparticles into the ferritin cage to mimic catalase and SOD activity.42,43
PbS quantum dots (QDs) can be prepared inside the ferritin cage for effective inhibition of growth of colorectal carcinoma cells via generation of ROS.45 The PbS-ferritin QDs are well tolerated by mice and no adverse effects were observed. Interestingly, the PbS-ferritin QDs were found to have NIR photoluminescence properties which are useful in combination with anticancer activity for cancer diagnostics, imaging and treatment.
The unique structure of the ferritin cage provides a suitable template for preparing small nanoclusters which have interesting fluorescence properties and can be utilized for cellular imaging. Nie and coworkers have developed an interesting strategy to assemble two gold clusters at the ferroxidase site of L-chain ferritin containing 2 H-chain subunits.36 The coupling interaction between the paired Au nanocluster gives enhanced fluorescence and is red shifted compared to the free clusters. The emission of the paired Au clusters can be tuned from green to far-red fluorescence. This emissive property was used for whole body imaging and it was found that the composite specifically targets kidney tissue (Fig. 2b). In a similar method, multiple Au clusters were prepared in H-chain ferritin which exhibits near-infrared (NIR) fluorescence with high quantum yield about 63% and used as a kidney targeting NIR imaging agent.37
2.2 Metal complexes in the ferritin cage
In addition to the preparation of nanomaterials, the ferritin cage provides suitable coordination environments for metal complexes for the generation of composites used in various applications. We have recently reported immobilization of organometallic [Pd(allyl)Cl]2 and [Rh(norbornadiene)Cl]2 complexes into the ferritin cage to promote catalytic C–C coupling and polymerization reactions and determined X-ray crystal structures of the composites.47,70,71 The ferritin cage has also been functionalized with several different metal complexes with the aim of controlling cellular functions, developing imaging agents and for drug delivery. The potent anticancer drug cisplatin and its analogues have been incorporated into the ferritin cage via an assembly/disassembly route (Fig. 3a).52,53 To enhance the loading efficiency, Pt particles were synthesized inside the ferritin cage by incubating K2PtCl4 in ammonia buffer (pH 8.5). The ferritin-encapsulated cisplatin has higher uptake efficiency than the naked drug which suggests that the system could overcome the cisplatin-related resistance problems. Receptor mediated endocytosis is thought to be responsible for the higher uptake of ferritin loaded platinum drugs. The cytotoxicity was found to be less than that of naked cisplatin due to limited release from the cage. However, the results indicate that ferritin has potential to be developed as a promising carrier for platinum-based anticancer drugs.52 | ||
| Fig. 3 Applications of the ferritin cage in drug delivery and imaging. (a) Incorporation of cisplatin into the ferritin cage for drug delivery.53 (b) Gadolinium chelate complex in the ferritin cage for magnetic resonance imaging (MRI).54 (c) Accommodation of zinc-phthalocyanine in the RGD-4C modified ferritin cage for targeted photodynamic therapy.51 | ||
A Gd(III)–chelate complex has been incorporated into the ferritin cage for the development of a high relaxivity contrast agent for magnetic resonance imaging (Fig. 3b).54 The cage can accommodate about 10 paramagnetic units and provide long-term stability with a low leaching rate. The resulting composite has 20 times higher longitudinal relaxivity than the naked chelate complex in aqueous medium. It is expected that the interaction between the protein interior cage and free exchangeable protons is responsible for the higher relaxivity. The Gd chelate has been loaded with curcumin, a polyphenolic compound for MRI guided treatment of cancer cells overexpressing ferritin receptors.56,57
In a similar manner, Mn-loaded ferritin has been developed as an MRI sensor of melanin formation in melanoma cells.58 The Mn2+ in the engineered ferritin cage exhibits high T2 relaxivity indicating its potential for use as an ultrasensitive T2 contrast agent.59
For targeted delivery, the surface of the ferritin cage was modified with the RGD-4C (Cys-Asp-Cys-Arg-Gly-Asp-Cys-Phe-Cys) peptide which selectively binds to integrin αvβ3, a tumor angiogenesis gene which is overexpressed in tumor cells.72 The RGD-4C-modified ferritin cage was then used to load a photoactive drug for selective delivery and controlled activation (Fig. 3c).51
Photoactive zinc hexadecafluorophthalocyanine (ZnF16Pc) has poor pharmacokinetics due to its aqueous insolubility. The solubility of this compound was found to be significantly improved by loading it into the RGD-4C modified ferritin cage. Despite the high loading (60 wt%) of ZnF16Pc, the overall particle size of apo-Ferritin remains the same. The photoactive ferritin nanocarrier has high tumor uptake efficiency in U87MG cells through RGD-integrin interaction and toxicity due to singlet oxygen generation when irradiated with red light. Recently, the ZnF16Pc loaded ferritin has been functionalized with folic acid as a tumor targeting ligand.73 The nanoconjugate enters cells via a folic acid receptor-mediated endocytosis pathway and suppresses tumor cell growth only in the presence of light while minimizing effects on normal cells. Interestingly, the conjugate exhibits PDT-stimulated suppression of lung tumor metastasis in vivo. Thus, the functionalized ferritin carrier has potential applications in targeted photodynamic therapy.
Recently, Ueno et al. used the apo-ferritin cage to deliver carbon monoxide (CO) into living cells (Fig. 4).48 CO in living cells acts as a signaling molecule to produce cytoprotective effects that counteract inflammation, proliferation and apoptosis.74 The organometallic [Ru(CO)3Cl2]2 complex used as a carbon monoxide releasing molecule (CORM) was incorporated into the cage of recombinant horse L-chain apo-ferritin (apo-rHLFr) (Fig. 4a). The resulting composite was found to have better activity than Ru(CO)3Cl(glycinate) (CORM-3), a typical CORM.48 The RuCO in the ferritin cage was substantially immobilized at the metal accumulation site and the 3-fold axis channel as determined by X-ray crystal structure analysis (Fig. 4b). The Fr-CORMs have ATR-IR CO stretching frequencies near 2040 and 2060 cm−1 indicating the presence of a cis-Ru(CO)2 coordination structure. The protein cage stabilizes the cis-Ru(CO)2 structure and reduces the rate of CO release compared to CORM-3. The slow release of CO is important for cellular applications as it is toxic to cells. The uptake of Fr-CORMs in living cells is 4-fold higher than that of CORM-3 and it has been confirmed that the composite releases CO inside the cell (Fig. 4c). Fr-CORMs were found to activate the nuclear factor κB (NF-κB), a transcriptional regulator of a number of pro-inflammatory and anti-apoptotic genes and considered as a potential therapeutic target of CO. The NF-κB activity of the non-toxic Fr-CORMs in the presence of tumor necrosis factor α (TNF-α) was found to be up to 4-fold higher relative to CORM-3. This shows the potential of a protein cage for controlling cellular functions.
 | ||
| Fig. 4 Carbon monoxide releasing ferritins. (a) RuCO incorporation into the ferritin cage.48 (b) Crystal structure of RuCO-ferritin showing the whole structure and Ru binding at the metal accumulation site. (c) Cellular uptake in the HEK293/κB-Fluc cell and activation of NF-κB by CO releasing RuCO-ferritin. (d) Crystal structure of photoactive MnCO-ferritin showing the whole structure and Mn binding at the metal accumulation site.49 | ||
After utilization of Fr-CORM, a photoactive CORM was constructed inside the ferritin cage for dose-regulated delivery in living cells.49 Photoactive Mn(CO)5Br was incorporated into the ferritin cage and characterized by X-ray crystal structure analysis (Fig. 4d). Although the CO ligands were not observed in the crystal structure, the ATR-IR was found to have three different CO stretching frequencies at 2028, 2011 and 1917 cm−1 corresponding to the fac-Mn(CO)3 coordination structure. Irradiation of visible light of 465 nm activates the MnCO-ferritin composite and releases CO in a time-dependent manner. The composite has uptake efficiency similar to CORM-1 (Mn2(CO)10) in HEK293/κB-Fluc cells and can be activated inside living cells in a controlled manner using light. The CO released from MnCO-ferritin in the HEK293/κB cell activates NF-κB in the presence of tumor necrosis factor α (TNF-α). In contrast, the naked CORM-1 does not show any activity despite similar uptake efficiency and large amounts of released CO relative to MnCO-ferritin. Therefore, the protein cage specifically stabilizes the photoactive MnCO moiety for light controlled activation of NF-κB.
In addition to applications in therapeutics and control of cellular functions, the ferritin cage can be engineered with neutral metal coordination moieties for biomimetic applications. The symmetric 3-fold axis channels formed by 3 monomer subunits of apo-ferritin are known to accommodate and transfer Fe ions into the cage. Such C3 axis channels are engineered to introduce a catecholamide derivative, N-(2-(2,5-dioxo-1H-pyrrol-1-yl)ethyl)-2,3-dihydroxybenzamide to mimic the enterobactin, a siderophore of Escherichia coli which is known as the strongest chelator of ferric ions (Fig. 5).50 In the presence of FeCl3, the modified ferritin exclusively produces the Fe(III)-tris-catecholato complex, as confirmed by the appearance of a characteristic LMCT band at 498 nm for the tris complex. The crystal structure analysis shows that Fe(III) forms an octahedral Fe(III)-tris-catecholato complex at the entrance of the 3-fold axis channel and blocks access of other molecules into the interior cavity. This work has broadened the scope of use of the ferritin cage for developing new biomimetic materials.
 | ||
| Fig. 5 (a) Molecular structure of Enterobactin. (b) Construction of an enterobactin analogue at the 3-fold channel of the apo-ferritin cage. | ||
2.3 Organic molecules in the ferritin cage
Several medicinally important organic molecules were incorporated into the ferritin cage to improve pharmacokinetics, activity and selectivity. Doxorubicin (Dox) was incorporated into the cage using a process involving disassembly in 8 M urea solution followed by reassembly after gradual removal of urea.63 The cage can accommodate 33 Dox molecules per apo-Fr without changing its overall structure and its size (12 nm) is considered to be optimal for an anticancer nano-medical agent to overcome the physiological barrier. Typical nanoparticles with sizes of about 100 nm are considered too large. The Dox loaded apo-Fr has excellent biocompatibility in vivo. The composite selectively delivers Dox molecules into tumor cells overexpressing TfR1 and does not require any additional functionalization to target cancer cells. The uptake of Dox-loaded ferritin in tumor cells was found to be 10-fold higher relative to free Dox and the composite has excellent antitumor activity compared to the clinically approved drug Doxil. In another study, the surface of the ferritin cage was modified with the RGD-4C (Cys-Asp-Cys-Arg-Gly-Asp-Cys-Phe-Cys) peptide which selectively binds to integrin αvβ3, a tumor angiogenesis gene which is overexpressed in tumor cells.64 Dox molecules were pre-complexed with Cu(II) and loaded into the ferritin cage via passive diffusion with a loading capacity up to ∼73% (w/w). Despite high loading, the ferritin cage structure was retained. The RGD modified ferritin cage loaded with Dox molecules is more effectively taken up by tumor cells due to the interaction between RGD-4C and integrins αvβ3 in U87MG cells. The Dox in the apo-Fr cage was released gradually and later identified after a long incubation time predominately in the nuclei where it interacts with DNA to induce cell death. The activity was found to be greater than that of free Dox. Similarly, an organic dye, methylene blue, was incorporated into the ferritin cage via an assembly/disassembly route for developing light-activated anticancer agents.66 In the presence of red light, the dye-encapsulated ferritin composite produces singlet oxygen which induces a cytotoxic effect in MCF-7 cells.Besides protein cages, there are a number of synthetic cages such as metal–organic frameworks (MOFs), zeolites, and polymersomes which are also available for controlling cellular functions.75–77 For example, engineered iron(III)-based metal–organic frameworks have been developed for drug delivery and imaging purposes.78 Similarly, a polymeric nanoreactor encapsulates the enzyme penicillin acylase for local and controlled production of antibiotics.79 The main advantage of the synthetic cages is that the cavity size can be easily tuned and also it can be functionalized for specific targeting ligands. However, the aqueous solubility, stability and biocompatibility remain an issue. In contrast, all such issues can be easily overcome by using the ferritin cage. The cage easily internalizes into cells. The preparation of the nanocomposites or metal functionalization using the ferritin cage can be achieved in a single step in buffer solution and does not require additional targeting or stabilizing ligands. Thus, the ferritin cage is a promising nano-platform for developing new biomaterials which are expected to provide the basis for new therapeutic and imaging applications.
3. Protein crystals
Protein crystals have been used in the determination of three-dimensional structures by X-ray crystallography since J. Kendrew first determined the three-dimensional protein structure of myoglobin in 1958.80 In the last two decades, protein crystals have been generally regarded as porous materials because protein crystals have highly ordered arrangements of protein monomers and offer a wide range of pore sizes (20–100 Å), high porosity (50–80%), and large pore surface areas.5,81 However, because protein crystals tend to be very fragile in buffer solutions except for crystallization buffer, cross-linked enzyme crystals (CLECs) and cross-linked protein crystals (CLPCs) have been exploited to improve the mechanical stability of crystals for application as solid porous materials for separation, storage, and enzymatic reactions.81,102,103 Recently, these efforts have expanded the potential for using protein crystals as nanomaterials for functionalizing metal ions, metal complexes and metal nanoparticles as well as organic compounds (Table 2).5 In this section, we provide a survey of recent developments in functionalization of solvent channels of protein crystals for the construction of hybrid protein biomaterials.| Protein | Composites | Methods | Functions | Ref. |
|---|---|---|---|---|
| a HEWL = hen egg white lysozyme. b KDPGal = 2-keto-3-deoxy-6-phosphogalactonate. c CCMV = cowpea chlorotic mottle virus. | ||||
| HEWLa | Au nanostructure | Soaking and chemical reduction (NaBH4) | Preparation of an Au nanostructure | 82 |
| Au NP | Soaking and autoreduction | Observation of the Au NP formation process | 83 | |
| Au NP | Soaking and autoreduction | Catalyst: p-nitrophenol reduction | 84 | |
| Au NP | Soaking and chemical reduction (NaOH) | Catalyst: p-nitrophenol reduction | 85 | |
| Ag nanostructure | Soaking and photo reduction | Preparation of an Ag nanostructure | 82 | |
| Ag NP | Soaking and chemical reduction (NaBH4) | Catalyst: p-nitrophenol reduction | 86 | |
| CoPt NP | Soaking and chemical reduction (NaBH4) | Magnetism | 87 | |
| Polypyrrole | Soaking and polymerization | Preparation of polypyrrole | 88 | |
| Carbon dots | Soaking | Luminescence | 89 | |
| CdS | Soaking | Fluorescence | 90 | |
| Rh3+ | Soaking | Observation of the metal accumulation process | 91 | |
| [Ru(benzene)Cl2]2 | Soaking | Catalyst: transfer hydrogenation | 92 | |
| [Ru(CO)3Cl2]2 | Soaking | Extracellular matrix for CO releasing | 93 | |
| Myoglobin | ZnP, Ru3O | Heme substitution, soaking | Electron transfer system | 94 |
| Ru complexes, fluorescent dye | Chemical modification before crystallization | Porous modification | 95 | |
| Ferritin | Pd ion | Immobilization | Observation of the metal accumulation process | 70 |
| Zn, organic ligand | Co-crystallization | Synthesis of a porous crystalline framework | 96 | |
| Dendrimer | Co-crystallization | Control crystal lattice structures | 97 | |
| Phthalocyanine and pyrene tetrasulfonic acid | Co-crystallization | 1O2 generation | 98 | |
| KDPGalb and FkpA Protein | — | Fusion proteins by the genetic method | Porous crystal | 99 |
| CCMVc | Avidin | Co-crystallization or soaking | Immobilization of functional molecules such as enzymes and Au NPs | 100 |
| Plastocyanin | PEG | Chemical modification | Structure characterization | 101 |
3.1 Crystal engineering of hen egg white lysozyme
Among the engineered protein crystals, hen egg white lysozyme (HEWL) crystals are most frequently exploited as molecular templates for synthesis of metal nanoparticles and immobilization of metal complexes.104 Because HEWL crystals are easily and abundantly obtained, we can investigate the properties and functions of the engineered crystals in detail as single crystals and in bulk. It has been reported that various metal ions and metal complexes are accumulated by coordination of amino acid residues on the surfaces of the solvent channels.91,105,106 In addition, we can obtain different polymorphic structures of HEWL crystals by changing crystallization conditions. Such polymorphic structures include tetragonal, orthorhombic and monoclinic structures (T-, O-, and M-HEWL), which have different pore sizes, shapes, and arrangements of amino acid residues on the channels (Fig. 6).107–109 Therefore, HEWL crystals are one of the most promising crystals used as solid materials. | ||
| Fig. 6 Crystal lattice structures and the major solvent channels of (a) tetragonal (T)-HEWL, (b) orthorhombic (O)-HEWL, and (c) monoclinic (M)-HEWL taken from the PDB codes of 103L, 1BGI, and 5LYM, respectively. | ||
 | ||
| Fig. 7 (a) Optical and (b) SEM micrographs of cross-linked lysozyme crystals containing chemically reduced Au nanoparticles. (c) TEM image of the thin fragments and corresponding EDX analysis and electron diffraction data. (d) Optical and (e) SEM micrographs of polypyrrole-cross-linked lysozyme crystals (PPy-CL-HEWL). (f) TEM image of the thin fragments of PPy-CL-HEWL. (g) Optical and (h) fluorescence micrographs of CL-HEWL (right), oxCD-CL-HEWL (middle), and CD-CL-HEWL (left). (i) TEM image of the thin fragments of CD-CL-HEWL. Reproduced from ref. 82, 88 and 89, with permission for (a–c), (d–f), and (g–i) from Wiley-VCH, the Royal Society of Chemistry and Wiley-VCH, respectively. | ||
Synthesis of polypyrrole (PPy) in CL-T-HEWL crystals was also accomplished (Fig. 7d–f).88 CL-T-HEWL crystals were soaked in the aqueous solution containing ammonium persulfate (APS) as an oxidant for pyrrole. Then the APS-CL-T-HEWL crystals were exposed to pyrrole vapor for synthesis of PPy in the solvent channels of the crystals. TEM images of the PPy-CL-T-HEWL crystals showed continuous striations with a thickness of 1.8 nm, which they attributed to PPy domains because the striation images were not observed without polymerization of pyrrole. PPy-CL-T-HEWL crystals have low conductivity with a resistivity of 104 Ω m, although CL-T-HEWL crystals have high resistivity (>107 Ω m).
Graphitic nanoparticles (carbon dots, CDs) were synthesized and confined within the solvent channels of the CL-T-HEWL crystals (Fig. 7g–i).89 CL-T-HEWL crystals were soaked in an aqueous solution including citric acid and ethylene diamine and then exposed to microwave radiation to form oxygen-containing graphite nanoparticles (oxCD-CL-T-HEWL) by inducing pyrolysis of incorporated molecules. Carbon dots within the crystals (CD-CL-T-HEWL) were synthesized by incubation of the oxCD-CL-T-HEWL in an aqueous solution of sodium borohydride (NaBH4). Fluorescence microscopy of CL-T-HEWL, oxCD-CL-T-HEWL and CD-CL-T-HEWL crystals indicated that the fluorescence intensities of the CD-CL-T-HEWL and oxCD-CL-T-HEWL crystals are dramatically increased and decreased, respectively, compared to the CL-T-HEWL crystal. It was found that fluorescence properties could be tuned by the addition of guest molecules or an auxiliary chromophore at acidic or neutral pH, because pH-induced donor–acceptor coupled excitation energy transfer provides white or green light-emitting crystals.
Yi Lu et al. have reported time-dependent formation of gold nanoparticles (Au NPs) using a single crystal of T-HEWL (Fig. 8a and b).83 The process of formation of Au NPs within the T-HEWL crystals was elucidated by auto-reduction of T-HEWL accumulating ClAuS(CH2CH2OH)2 (Au(I)) using transmission electron microscopy (TEM) and X-ray crystallography. Because the rate of auto-reduction of Au(I) is slow, it is possible to observe the process of formation of Au NPs. The time-dependent observation of Au NP formation could provide a means to elucidate biomineralization mechanisms and metal cluster formation mechanisms on the surfaces of proteins. Catalytic reactions for reduction of p-nitrophenol were also evaluated using Au NPs synthesized in the T-HEWL crystals with NaBH4.84 Smaller Au NPs (2.2 nm) exhibited higher catalytic activities compared to the T-HEWL crystals with larger Au NPs (>10 nm).
 | ||
| Fig. 8 Time-dependent growth of Au NPs in the single crystal of HEWL. (a) Optical images of single crystals of HEWL with Au(I) ions on different days of growth. (b) Corresponding TEM images. Reproduced from ref. 83 with permission for (a and b) from Nature publishing group. | ||
Fluorescent quantum dots were prepared in the single HEWL crystals by co-crystallization of lysozyme and Cd(II) ions in the presence of sodium sulfide.90 The hybrid crystals were found to emit red fluorescence with a peak centered at ∼700 nm under fluorescein isothiocyanate illumination. In the absence of sodium sulfide, the crystals containing Cd(II) ions are colorless and non-fluorescent, indicating that quantum dots of CdS are formed in the single crystal of lysozyme. In addition, when the CdS was prepared in aqueous solution in the presence of the lysozyme monomer, a low level of red fluorescence was observed, and red fluorescence emission was found to disappear after dissolution of the hybrid crystals containing CdS. These results show that only preparation of CdS inside the crystals exhibits enhancement of fluorescence due to the unique environments provided by the solvent channels of the crystals.
Ueno et al. synthesized magnetic CoPt NPs within the solvent channels of HEWL crystals with different sizes and compositions using three different crystal systems of HEWL (Fig. 6 and 9).87 T-HEWL, O-HEWL and M-HEWL crystals were cross-linked with glutaraldehyde to maintain the crystal lattice during synthesis of CoPt-NPs. Cross-linked HEWL crystals (CL-O-HEWL, CL-T-HEWL, and CL-M-HEWL) were soaked in buffer solution containing Co(II) and Pt(II) ions. The CoPt NPs were synthesized by reduction of the CL-HEWL crystals containing Co and Pt ions with sodium borohydride. The average particle sizes of CoPt NPs synthesized in CL-HEWL crystals were determined using TEM. Alignment of CoPt NPs in the solvent channels of the CL-HEWL crystals was observed (Fig. 9b–d). The composition of Co and Pt in CoPt NPs was analyzed by determining the X-ray fluorescence of CoPt NPs in CL-O-HEWL, CL-T-HEWL and CL-M-HEWL crystals. The results show that the ratios of Co and Pt atoms in CoPt NPs are 7.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :92.3, 3.8:96.2, and 6.3:93.7, respectively. The composition of CoPt NPs in the CL-HEWL would be affected by pre-organization of Co(II) and Pt(II) ions bound on the surface of the solvent channels of the HEWL crystals. The magnetic properties of CoPt·CL-HEWL crystals were examined using a superconducting quantum interference device (SQUID) magnetometer. CoPt·CL-O-HEWL has the highest coercivity value (4600 Oe) among the three CoPt·CL-HEWL crystals (with 1600 and 2900 Oe for CoPt·CL-T-HEWL and CoPt·CL-M-HEWL, respectively). The order of the coercivity of CoPt NPs within the crystals is generally proportional to the content of Co atoms of the CoPt NPs as determined in the previous report. These results indicate that the physical properties of inorganic metal nanoparticles prepared in the solvent channels can be tuned by the crystal lattice.
:92.3, 3.8:96.2, and 6.3:93.7, respectively. The composition of CoPt NPs in the CL-HEWL would be affected by pre-organization of Co(II) and Pt(II) ions bound on the surface of the solvent channels of the HEWL crystals. The magnetic properties of CoPt·CL-HEWL crystals were examined using a superconducting quantum interference device (SQUID) magnetometer. CoPt·CL-O-HEWL has the highest coercivity value (4600 Oe) among the three CoPt·CL-HEWL crystals (with 1600 and 2900 Oe for CoPt·CL-T-HEWL and CoPt·CL-M-HEWL, respectively). The order of the coercivity of CoPt NPs within the crystals is generally proportional to the content of Co atoms of the CoPt NPs as determined in the previous report. These results indicate that the physical properties of inorganic metal nanoparticles prepared in the solvent channels can be tuned by the crystal lattice.
 | ||
| Fig. 9 (a) Schematic drawing of the preparation of CoPt-NPs in a solvent channel of a HEWL crystal. TEM images and particle size distribution of (b) CoPt·CL-O-HEWL, (c) CoPt·CL-T-HEWL, and (d) CoPt·CL-M-HEWL. The black dots are CoPt NPs and the white dotted lines (insets) provide a guide. | ||
 | ||
| Fig. 10 (a) Schematic representation of the preparation of Ru(benzene)·CL-HEWL and catalytic transfer hydrogenation. Crystal structures in a lattice structure of (b) Ru(benzene)·CL-T-HEWL and (d) Ru(benzene)·CL-O-HEWL and active site structures of (c) Ru(benzene)·CL-T-HEWL and (e) Ru(benzene)·CL-O-HEWL. | ||
HEWL crystals immobilized with organometallic Ru(CO) complexes can function as an extracellular matrix (ECM) for CO release.93 The [Ru(CO)3Cl2]2 complex (CORM-2) was immobilized in the solvent channels of CL-T-HEWL crystals in an effort to store and release CO gas by coordination of amino acid residues such as His, Asp and Lys as observed from the X-ray crystal structure of Ru(CO)·CL-T-HEWL (Fig. 11). The number of Ru complexes in the crystals was found to be 10 per HEWL monomer. The IR spectrum of Ru(CO)·CL-T-HEWL exhibits four bands at 2055, 2025, 1981, and 1940 cm−1. A pair of IR bands at 2055 and 1981 cm−1 was assigned to cis-Ru(CO)2 complexes coordinated with His15. The other pair of bands at 2025 and 1940 cm−1 was expected for cis-Ru(CO)2 complexes ligated to carboxylate of an Asp residue. The CO release phenomenon was examined in a myoglobin assay based on conversion of deoxy-myoglobin to carbonyl-myoglobin. The half life (t1/2) value of CO release from Ru(CO)·CL-T-HEWL was found to be increased 10-fold longer than CORM-2. The amount of CO released from Ru(CO)·CL-T-HEWL was found to be 0.38 per Ru. The NF-κB activity of Ru(CO)·CL-T-HEWL in the presence of TNF-α was found to be increased by 20% compared to the control.
 | ||
| Fig. 11 (a) Schematic representation of the preparation of Ru(CO)·CL-HEWL. (b) Crystal structure in a lattice structure and coordination structure in the vicinity of Asp18 of Ru(CO)·CL-HEWL. (c) CO release in the vicinity of cells and activation of NF-κB. | ||
3.2 Crystal engineering of myoglobin
Sperm whale myoglobin is crystallized with ammonium sulfate as a precipitant. The myoglobin crystal belongs to the space group P6 and has hexagonal pores with a diameter of 40 Å. This pore can be employed as a reaction space and metal accommodation space within the crystals.95 A Zn porphyrin (ZnP) and a Ru3O cluster were found to be accumulated on the specific sites in the solvent channels of a single crystal of Mb by the replacement of heme and coordination of His48 and His12 to construct an artificial photo-induced electron transfer system (Fig. 12).94 Methyl viologen (MV) soaked in the crystals was employed as a mediator between the ZnP and the Ru3O cluster. This functionalized Mb crystal provides a longer half-life of the charge-separated state which is 2800-fold higher than that of the same system in organic solvent. This result suggests that the protein crystal can accumulate functional molecules at specific sites in the solvent channels to develop unique activity.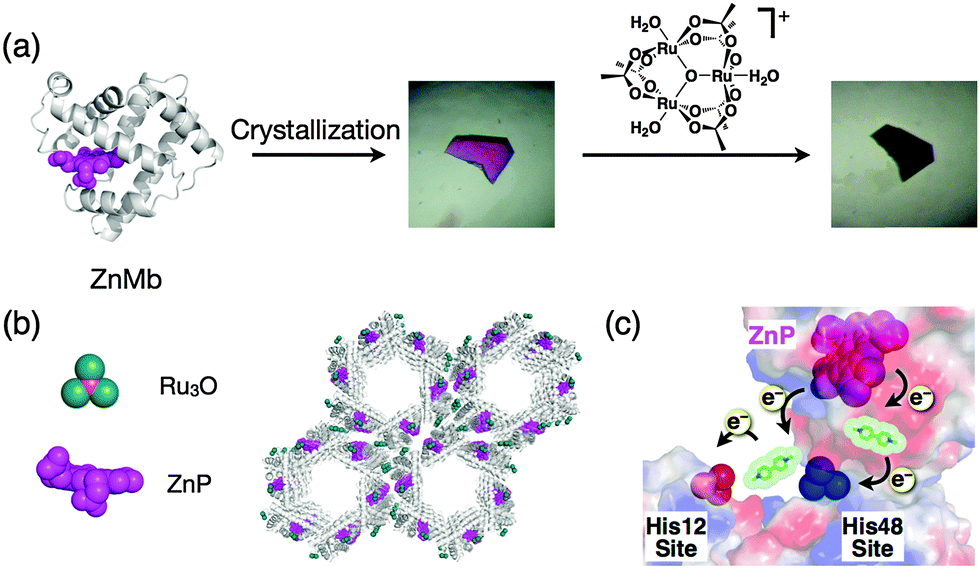 | ||
| Fig. 12 (a) Construction of a photoinduced electron transfer system in the ZnP substituted Mb crystal. (b) Crystal structure of the Ru3O soaked ZnMb crystal. (c) Schematic representation of methyl viologen mediated electron transfer between ZnP and Ru3O in ZnMb crystals. | ||
3.3 Crystal engineering of ferritin
Ferritin (Fr) is a spherical protein cage composed of 24 subunits with an interior space 8 nm in diameter. Apo-Fr has been utilized as a protein container for deposition of metal nanoparticles and for the incorporation of metal complexes.32,34 Recently, crystal engineering using ferritin has been developed to construct novel assembly structures. Ueno and co-workers elucidated the mechanism for accumulation of Pd ions on the interior surface of recombinant horse L-chain apo-ferritin (apo-rHLFr) using X-ray crystal structural analysis of a series of apo-rHLFr containing Pd ions (Fig. 13a).70 Apo-rHLFr containing different amounts of Pd ions (50, 100, and 200 equivalences vs. apo-rHLFr) was crystallized. X-ray crystal structures of these Pd·apo-rHLFr composites show that Pd ions are deposited at specific binding sites located at the accumulation sites and 3-fold channels. At the accumulation center, Pd ions are selectively bound to Cys48 and His49 with several different binding modes at an early stage of Pd binding. The conformational changes of His49, Glu45 and Arg52 stabilize and promote Pd(II) binding at the accumulation center. These interactions would promote the natural accumulation reactions such as biomineralization and metal cluster formation in protein scaffolds. | ||
| Fig. 13 (a) Accumulation process of Pd ions on the interior surface of apo-rHLFr. (b) Structure of the 3-fold channel of the ferritin mutant. (c) The bcc packing of the bdh-Zn-ferritin lattice mediated by coordination of bdh2− with Zn bound at 3-fold channels. (d) Closeup view of the interface of two ferritin molecules, bdh2− ligand bridge ferritin molecules. The structure was taken from PDB code 5CMR. | ||
Tezcan and co-workers have established the construction of protein supramolecules from 1D protein nanotubes to 3D protein crystals by metal-mediated protein assemblies using engineered cytochrome cb562 and ferritin.110,111 A 3D porous crystalline framework has been constructed recently by using the metal-directed protein assembly using ferritin, which was engineered at the 3-fold channel (Fig. 13b–d).96 Thr122 in the exterior of the 3-fold axis channel was replaced with His to introduce a Zn coordination site. In addition, original cysteine residues of Cys90, Cys102, and Cys130 were replaced with either Glu or Ala to prevent Zn binding at these sites. 3D porous frameworks are formed by coordination of Zn2+ with the mutated ferritin in the presence of the bidentate ligand, benzene-1,4-dihydroxamic acid (H2bdh) as well as metal organic frameworks (MOFs) which are composed of metal ions and organic ligands. The crystal structure of the framework has a different crystal lattice structure (I432) from the crystal produced in the presence of only ferritin and Zn ions (F432), and ferritin molecules were found to be connected by coordination of bidentate ligands to Zn ions (Fig. 13c and d). The self-assembling structure with metal coordination provides highly porous frameworks with a solvent content of 67%. This work indicates an opportunity to generate template proteins based on crystalline biomaterials for useful applications.
Kostiainen and co-workers have recently reported functionalization of a ferritin crystal by co-crystallization of dendrimers or organic dyes.97,98 The electrostatic self-assembly of apo-Fr and poly(amidoamine) (PAMAM) dendrimers provides two-component crystalline materials. The crystalline lattice structures of the co-crystals are affected by ionic strength and dendrimer generation. Two crystal symmetries (fcc and hcp) are allowed using apo-Fr and the dendrimer. The lattice structures also depend on the size of the dendrimer and the ratio of the dendrimer and apo-Fr.77 The organic dyes were also co-crystallized with apo-Fr. The supramolecular complex formed between cationic Zn phthalocyanine and anionic pyrene acts as molecular glue to crystallize apo-Fr via electrostatic interactions. The small-angle X-ray scattering (SAXS) analysis of the crystal shows the face-centered cubic (fcc) structure. The adsorption spectrum of the hybrid crystals indicates that the complex of phthalocyanine and pyrene is maintained in the crystals. The hybrid crystals can generate highly reactive 1O2 which is useful for various applications such as photodynamic therapy, water treatments, diagnostic arrays and as an oxidant in organic synthesis.98
These results indicate that apo-Fr is useful for engineering crystals as well as in vivo applications due to its high stability. The engineered Fr crystals can be used to create functional solid materials because ferritin crystals can utilize both the interior of the cage and the solvent channels of the crystals.
3.4 Crystal engineering of other proteins
Artificial protein crystals have been designed by introducing and modifying functional moieties at the interface of proteins, such as fusion of two protein domains, which naturally form oligomeric proteins for the formation of novel protein crystals as reported in our recent review.5 Co-crystallization of two different proteins provides heterogeneous crystalline assemblies integrating different functions into nanostructures.100 Protein–polymer conjugates were also crystallized and the protein–polymer interactions were investigated.101Yeates and co-workers have reported novel protein caged assemblies of 750 kDa with a large interior space of about 130 Å by fusion of trimeric Escherichia coli 2-keto-3-deoxy-6-phosphogalactonate (KDPGal) aldolase and dimeric N-terminal domain E. coli FkpA protein (Fig. 14a–c).99 The fusion protein is composed of KDPGal aldolase at the N-terminal domain, the N-terminal domain of FkpA at the C-terminal domain and a four residue alpha helical linker. The fusion protein designed by computational modeling was crystallized after a prolonged incubation time. The crystal structure showed that the atomic structure is in agreement with the modeled structure within a 1.2 Å root-mean-square deviation. In the crystal, large voids are formed with high solvent content (∼82%) as well as porous organic molecules and metal organic frameworks. These specifically designed highly porous protein crystals can be used to immobilize large molecules such as proteins for X-ray crystal structure analysis.
 | ||
| Fig. 14 (a–c) Crystal structure of the fusion protein of aldolase-three-helix cubic protein. (a) Four complete cages of the ATC-HL3 cage are packed within a unit cell. (b) The cube-shaped cage in the crystal. (c) The packing alignment of cages in the crystal produces a highly porous protein lattice. (d) Pre- and post-functionalization of CCMV-avidin crystals thorough biotin–avidin interaction. Reproduced from ref. 99 and 100 with permission from Nature publishing group. | ||
Kostiainen et al. constructed virus crystals including different functional materials, such as proteins, dendrimers, and metal nanoparticles.100 Cowpea chlorotic mottle virus (CCMV) was crystallized in the presence of avidin, to generate an electrostatically assembled composite with a non-close-packed body centered cubic structure (Fig. 14d). The multicomponent crystal was modified with biotin-conjugated fluorescein, horseradish peroxidase, and gold nanoparticles to incorporate these functional molecules into the crystals using avidin-biotin interactions. The multicomponent crystals were also prepared by co-crystallization of CCMV and pre-modified avidin with biotinylated functional molecules. The functionalized crystals have 2 orders of magnitude higher enzymatic activity (0.13–0.15 μM s−1) than the corresponding free enzymes in supernatant solution which are obtained after washing crystals (∼0.007 μM s−1). Furthermore, only crystalline avidin-HRP CCMV crystals show high enzymatic activity compared to amorphous HRP without CCMV. These results show that active enzymes are accumulated into the crystals and that this method can be applied to several biotinylated molecules for applications in preparation of crystalline materials having catalytic, optical and magnetic functions.
Crowley et al. reported structural characterization of the β-sheet protein plastocyanin (Pc) conjugated with a single poly(ethylene glycol) (PEG) 5000 molecule by size exclusion chromatography (SEC), NMR spectroscopy, and X-ray crystallography.101 The crystal structure refined at 4.2 Å resolution has a highly porous structure with high solvent content (80%), in which the PEGylated proteins form double-helical assemblies, although the electron densities corresponding to the PEG chains are not observed due to the disorder. The volume of the PEG domain in the crystals was estimated to be within 10% of the calculated random coil PEG 5000. The NMR studies show that the PEG domains are not influenced by the proteins, suggesting that the PEG domains have minimal protein interactions.
These engineered protein crystals with novel crystal assembling structures have significant potential to provide large spaces within crystals in the development of applications in solid functional materials to accumulate large molecules such as enzymes and proteins.
4. In vivo three dimensional protein crystals
As mentioned in the previous section, there is growing interest in functionalization of protein crystals. However, it remains challenging to establish large scale or more conventional systems for tailoring protein crystals with various functions. It is well known that certain proteins can be crystallized in living cells, such as peroxidase in peroxisomes, insulin in secretory granules, and polyhedrin crystals for generating functional solid-state catalysts, generating storage vessels, and encapsulating viruses, respectively.112–114 These protein crystals are spontaneously formed in living cells with high concentrations of the proteins in restricted locations. These crystals tend to be quite small compared to crystals prepared in vitro because growth of protein crystallization is limited to the size of living cells. Therefore, in vivo protein crystals have not been used for X-ray crystal structural analysis until recently due to radiation damage and difficulties in focusing on microcrystals.115 Enhanced techniques in micro-X-ray crystallography allow us to solve these problems by using microfocus X-ray beamlines and X-ray free-electron lasers.115 In 2007, Coulibaly et al. reported the first crystal structure of the in vivo protein crystal polyhedra, which forms in silkworms infected by cypoviruses (cytoplasmic polyhedrosis virus, CPV).116 Several in vivo protein crystals, including polyhedra and cathepsin B from Trypanosoma brucei (CatB), are produced in insect cells (Fig. 15).114,116,117 Structures of insecticidal Cry3A toxin in Bacillus thuringiensis, viral spindles, and coral fluorescent protein have been determined by microcrystal X-ray crystal structure analysis.114,118–120 In this section, recent progress in in vivo protein crystallization and in vivo crystal engineering toward the development of functional solid materials is described. | ||
| Fig. 15 In vivo three dimensional protein crystals. (a) Polyhedral crystals produced in Sf21 insect cells. (b) Scanning EM image of Sf9 insect cells infected with cathepsin B from Trypanosoma brucei (CatB) virus. (c) Phase contrast light micrograph of Bt cells. The dark rectangular shapes correspond to Cry3A toxin crystals. Reproduced from ref. 117 with permission for (b) from Nature publishing group. (c) was reproduced from ref. 118. Copyright (2014) National Academy of Science, USA. | ||
4.1 In vivo protein crystallization
The crystal structures of several in vivo protein crystals have recently been determined using synchrotron X-ray microbeam- and X-ray free-electron laser (XFEL)-based crystallography techniques.114–116,118–120 Coulibaly et al. reported the crystal structures of polyhedral crystals (PhCs), which are crystals of polyhedrin monomers (PhMs) produced in insect cells infected by cypoviruses (CPVs) and nuclear polyhedrosis viruses (NPVs).116,121 During crystallization in the cell, polyhedra embed the virus particles. The viruses embedded in PhCs are protected against harsh conditions. The crystal structure of CPV PhCs shows that a trimer of PhMs tightly organized crystalline structures, in which the H1-helices are located at the N-terminus of PhMs, interact with neighboring PhMs. The dense pack of trimer structures forms narrow solvent channels in the crystals (Fig. 16a–c). The structures of wild-type and recombinant NPV PhCs were also determined by X-ray crystallography.121,122 The recombinant structure shows that molecular organization of NPV polyhedrin is different from CPV polyhedrin. The assembling structures of baculovirus PhCs are stabilized by disulfide bonds and domain-swapping of N-terminal regions (Fig. 16d–f). Therefore, the N-terminal domains play different roles in CPV and NPV PhCs for stabilization of the crystalline assembly. | ||
| Fig. 16 Crystal structures of CPV polyhedra (a–c) and NPV polyhedra (d–f). Monomer (a and d) and trimer (b and e), and molecular assembling of the polyhedrin trimer in a unit cell (c and f) of CPV PhCs, and NPV PhCs taken from PDB ID: 2OH6, 3JVB, respectively. | ||
Coulibaly et al. also reported a crystal structure of the spindle produced by insect poxviruses. Spindles are crystalline assemblies of fusolin protein that enhance the virulence of the viruses and the insecticidal activity of unrelated pathogens.119 To understand the mechanism of enhancement of the virulence and assembly of spindles for ultra-stable crystalline materials, the spindle structure was determined by X-ray crystallography. The crystal structure of spindle from Melolontha melolontha EV (MMEV) shows that the spindles are stabilized by cross-linking of the C-terminal molecular arm of fusolin as well as the H1 helix of CPV PhCs and disulfide bonds between fusolin dimers to form the 3D network through the entire group of crystalline assemblies of spindles.
X-ray free-electron lasers (XFELs) are also utilized for crystal stucture analysis of in vivo protein crystals. The crystal structure of CatB grown in insect cells was determined using XFELs and refined to 2.1 Å.114 The native structure of CatB includes the pro-peptide and carbohydrate, which were not observed in the crystals of recrystallized CatB. This crystal structure analysis of in vivo protein crystals has potential for use in estimating the in vivo dynamic reactions of proteins, such as glycosylation, and phosphorylation, among others.
Very recently, crystal structure analysis of in vivo protein crystals within cells has been developed using microfocus beamlines with synchrotron and X-ray free-electron laser beam sources.118,120,123 Whole cells bearing CPV PhCs and engineered fluorescent protein variants were spread on a mesh loop, cooled, and irradiated at X-ray microfocus beamlines.120,123 The structure of CPV PhCs collected within cells indicates that there is no significant difference from isolated crystals. Sawaya and co-workers reported a crystal structure of Cry3A toxin produced in Bacillus thuringiensis (Bt) determined in an in vivo diffraction study using XFELs.118 The structure of Cry3A existing in living cells was determined to a resolution of 2.9 Å. The method of in vivo diffraction data collection does not require purification of the in vivo protein crystals and thus avoids destabilization of the crystals that can occur when the crystals are isolated from cells. In the future, in vivo chemical reactions could be analyzed by in vivo X-ray crystallography of in vivo protein crystals within living cells.
Miyawaki et al. have recently reported in vivo crystallization of a variant of coral fluorescent protein, Kikume Green-Red (KikGR), in HEK293 cells.120 The KikGR variant was transfected into HEK293 and the crystallization process was observed using confocal microscopy. The time-dependent observation indicated that the crystals grew to micro-sized crystalline assemblies over the course of several minutes in living cells after 2–5 days of transfection (Fig. 17). TEM images and biochemical experiments indicate that the KikGR variant is crystallized within the cytoplasm and then the crystal is incorporated into the lysosome as a result of an autophagic process.
 | ||
| Fig. 17 (a) Transmission (T), fluorescence (FL) and merged images of the HEK293 cell bearing a crystal of a coral fluorescent protein, KikGR, variant. (b) Crystallization process of the KikGR variant revealed by long-term imaging. Differential interference contrast (DIC) and fluorescence (FL) images of HEK cells at the indicated times. Reproduced from ref. 120 with permission from Cell Press. | ||
4.2 In vivo crystal engineering
The most prominent example of generation of in vivo protein crystals is the polyhedra produced in baculovirus-infected insect cells. CPV produces a protein known as polyhedrin in the replication cycle in insect cells to embed the viruses in spheres with a diameter of 75 nm in the crystals. The virus particles embedded in PhCs are resistant to dehydration, freezing and enzymatic degradation for several years because PhCs can tolerate harsh chemical treatments including incubation in concentrated urea, acid and detergents.116 Therefore, a PhC is a candidate for development as a crystalline solid material because the PhC is easily and abundantly crystallized in insect cells and has high stability.Mori et al. reported encapsulation of exogenous proteins into recombinant PhCs instead of CPV by co-expression of the exogenous protein and PhM.124,125 The H1, N-terminal fragment of the PhM can function as a polyhedrin recognition signaling moiety which leads to the incorporation of exogenous proteins or enzymes because the H1-helix tightly interacts with neighboring PhMs as well as VP3 of CPV turret protein (Fig. 16a–c).124,125 Fusion proteins consisting of enhanced fluorescent protein (EGFP) fused with the H1-helix fragment of the PhM (H1-EGFP) or VP3-fragment of CPV (EGFP-VP3) were co-infected to prepare the composites of PhCs which include H1-EGFP or EGFP-VP3 (Fig. 18). Confocal laser scanning microscopy of the crystals indicates that the green fluorescence of EGFPs is distributed more close to the surface than to the core of the crystals. In addition, EGFP and Discosoma sp. red fluorescent protein (DsRed) are co-immobilized in one PhC by using the H1 and VP3 fragments. This technique was applied to immobilize various cytokines, such as FGF, LIF, and EGF in PhCs.126,127 These immobilized cytokines retain their biological activities, which include promotion of fibroblast growth and proliferation of mouse embryonic and induced pluripotent stem cells by slow release of the immobilized proteins.
 | ||
| Fig. 18 Encapsulation of H1-EGFP into CPV PhC by co-infection and co-expression of CPV PhMs and H1-EGFP. | ||
Ueno et al. reported one-pot preparation of PhCs with protein kinase C (PKC) βII, a phosphorylation enzyme, by co-expression of PKC βII and PhMs, which is mutated for dissolution at optimum pH and retention of PKC activity after storage under drying. These modifications are expected to expand the application of the PhC as a solid material while the enzymes are retained at optimal pH (Fig. 19).128 The crystal structure of WTPhCs indicates that the side chain of Arg13 at the tip of the H1-helix is expected to contribute to the robust character of the crystals by the formation of intermolecular hydrogen bonds. Two mutant crystals were designed, in which Arg13 is replaced with Ala and Lys, R13APhMs and R13KPhMs, respectively. H1-PKC·R13APhCs and H1-PKC·R13KPhCs both release large amounts of H1-PKC molecules at pH 8.5 upon dissolution of PhCs, although H1-PKC could not be released from WTPhCs. Phosphorylation reactions were examined to evaluate the enzymatic reactions. The stabilization of H1-PKC within the crystals was evaluated by determining enzymatic activity after incubation by air-drying for 1 week at 25 °C (Fig. 19g). These results indicate that H1-PKCs immobilized in the crystals retain the enzyme activity after incubation under harsh conditions, although free-PKC does not retain activity under the same conditions. These results show that exogenous enzymes with an H1 helix can be incorporated by co-expression into insect cells and the mutant crystals can release the H1-PKC at pH 8.5 by dissolution of the crystals. H1-PKC has higher tolerance to drying when incorporated into the crystals compared to free PKC.
 | ||
| Fig. 19 Schemetic representation of (a) in vivo preparation of H1-PKC·PhC. (b) The interaction of the H1 helix with neighboring polyhedrin molecules. Magnified structures in the vicinity position 13 of (c) WTPhC, (d) R13APhC, and (e) R13KPhC. Hydrogen bonds are indicated with yellow dotted lines. (f) Schematic representation of release of H1-PKC by dissolution and their enzymatic reaction. (g) PKC activity of H1-PKC·PhCs and recombinant free PKC β-II after air drying for 1 week at pH 8.5. | ||
It has been demonstrated that the surface of PhCs can be modified with carbohydrate (Fig. 20a).129 Cys mutants, in which original Cys residues were replaced with Ala and one or two Cys residues, were introduced on the surface of the crystals. The introduced Cys residues were modified with propargyl maleimide and then the acetylene moieties were modified with Lewis X (LeX)-azide via the copper-catalyzed azide alkyne cycloaddition reaction. The reaction of LeX-modified PhCs with Alexa Fluor 488-conjugated antibody against stage-specific embryonic antigen 1 (SSEA-1) was performed for 10 h at 4 °C. Confocal laser scanning microscopy of the composite shows that Cys-mutant PhCs react with the antibody more effectively than WTPhCs. The surface modification of protein crystals is expected to provide attractive materials for applications involving extracellular matrices, biosolid catalysis, and drug delivery systems.
 | ||
| Fig. 20 (a) Schematic representation of surface modification of PhC with LeX moieties. The cystein residues were modified with propargyl maleimide then LeX-azide via thiol-maleimide chemistry and the copper-catalyzed azide alkyne cycloaddition, respectively. The LeX modifeid PhC was reacted with antibody. (b) Immobilization of Ru carbonyl complexes into PhC with a His-tag at the C-terminus. | ||
A PhC also reacted with organometallic Ru carbonyl complexes in an effort to construct CO-releasing extracellular matrices (Fig. 20b).130 Introduction of a hexahistidine tag (HT) to the C-terminus allows the crystal to accumulate Ru carbonyl complexes which are twice the size of WTPhCs, and release CO gas 3-fold more effectively than WTPhCs including Ru carbonyl complexes. The HT-PhC which includes Ru carbonyl complexes (Ru·HT-PhC) releases 6.2 ± 0.9 equiv. per PhM with a half-life (t1/2) of 27.7 ± 1.6 min. The value is about 4-fold greater than that of the original CORM-2 (6.9 ± 2.8 min). The performance of a Ru·HTPhC as an extracellular scaffold was evaluated by measuring the activity of nuclear factor kappa B (NF-κB) in living cells because CO gas is known to activate NF-κB in the presence of tumor necrosis factor α (TNF-α). Ru·HTPhCs showed a 72% increase of NF-κB activity compared to WTPhCs. The results indicate that the PhC can immobilize organometallic complexes to release the small molecules without degradation of the crystals. The hybrid crystals will be investigated for the development of solid functional materials for applications such as catalysis, metallodrugs, and bioimaging because organometallic complexes have various functions.
5. Conclusions and future perspectives
The design of protein assemblies to construct protein-based hybrid materials with metal ions, metal complexes, nanomaterials and proteins has recently become a growing field with the aim of providing novel functions and mimicking natural functions in materials science and nanotechnology. This feature article summarizes recent development of strategies for functionalization of protein assemblies based on the ferritin cage and protein crystals for applications such as catalysis, magnetism, optics, imaging, and drug delivery both in vivo and in vitro systems. Protein assemblies provide confined environments for accumulation of functional molecules. Self-assembled protein cages containing metal complexes and inorganic materials have been recently utilized in cellular environments for delivery of CO as a biological gas and delivery of drugs due to their high biocompatibility and stability. Furthermore, protein cages have been employed as reaction vessels to incorporate multiple catalytic centers such as enzymes for cascade-type reactions in larger internal spaces functioning as natural micro-compartments. In the future, catalytic cascade reactions in cellular environments using protein assemblies incorporating metal complexes and enzymes will be investigated.Protein crystals have also been recently developed not only in structural biology but also in materials science. Cross-linked protein crystals encapsulating metal ions, metal complexes and nanomaterials immobilized in solvent channels by coordination of amino acid residues and/or chemical modifications can function as solid biomaterials with catalytic, magnetic and optical properties. These efforts in protein crystal engineering have been expanded to in vivo protein crystallization. One of the advantages of using in vivo protein crystals as solid materials is that it is possible to prepare tailored protein crystals in large scale or conventional preparations because the crystals are spontaneously formed in living cells. Thus, design of confined chemical environments in protein cages and crystals has contributed to the development of new biohybrid materials.
Abbreviations
| Hsp | Heat shock protein |
| Dps | DNA-binding protein for starved cells |
| CCMV | Cowpea chlorotic mottle virus |
| SOD | Superoxide dismutase |
| ROS | Reactive oxygen species |
| DCFH-DA | 2,7-Dichlorofluorescein diacetate |
| TfR1 | Transferrin receptor 1 |
| TMB | 3,3,5,5-Tetramethylbenzidine |
| DAB | Di-aza-aminobenzene |
| QDs | Quantum dots |
| NIR | Near-infrared |
| MRI | Magnetic resonance imaging |
| RGD-4C | Cys-Asp-Cys-Arg-Gly-Asp-Cys-Phe-Cys peptide |
| PDT | Photodynamic therapy |
| CORM | Carbon monoxide releasing molecule |
| ATR | Attenuated total reflection |
| NF-κB | Nuclear factor κB |
| TNF-α | Tumor necrosis factor α |
| Dox | Doxorubicin |
| CLECs | Cross-linked enzyme crystals |
| CLPCs | Cross-linked protein crystals |
| HEWL | Hen egg white lysozyme |
| PPy | Polypyrrole |
| APS | Ammonium persulfate |
| CD | Carbon dot |
| NaBH4 | Sodium borohydride |
| Au NPs | Gold nanoparticles |
| TEM | Transmission electron microscopy |
| SQUID | Superconducting quantum interference device |
| ECM | Extracellular matrix |
| ZnP | Zinc porphyrin |
| MV | Methyl viologen |
| H2bdh | Benzene-1,4-dihydroxamic acid |
| MOF | Metal organic framework |
| PAMAM | Poly(amidoamine) dendrimer |
| SAXS | Small-angle X-ray scattering |
| Fcc | Face-centered cubic |
| KDPGal | 2-Keto-3-deoxy-6-phosphogalactonate |
| CPV | Cytoplasmic polyhedrosis virus |
| CatB | Cathepsin B |
| XFEL | X-ray free-electron laser |
| PhC | Polyhedra crystal |
| PhM | Polyhedrin monomer |
| NPV | Nuclear polyhedrosis virus |
| Bt | Bacillus thuringiensis |
| KikGR | Kikume green-red |
| EGFP | Enhanced green fluorescent protein |
| DsRed | Discosoma sp. red fluorescent protein |
| PKC | Protein kinase C |
| SSEA-1 | Stage-specific embryonic antigen 1 |
Acknowledgements
Parts of this work were supported by the Funding Program for Next Generation World-Leading Researchers (for T. U.) and Grant-in Aid for Scientific Research (for S. A.) from Ministry of Education, Culture, Sports, Science and Technology, Japan.Notes and references
- M. Uchida, M. T. Klem, M. Allen, P. Suci, M. Flenniken, E. Gillitzer, Z. Varpness, L. O. Liepold, M. Young and T. Douglas, Adv. Mater., 2007, 19, 1025–1042 CrossRef CAS.
- Coordination Chemistry in Protein Cages: Principles, Design, and Applications, ed. T. Ueno and Y. Watanabe, John Wiley & Sons, New Jersey, 2013 Search PubMed.
- B. Maity, K. Fujita and T. Ueno, Curr. Opin. Chem. Biol., 2015, 25, 88–97 CrossRef CAS PubMed.
- H. Inaba, S. Kitagawa and T. Ueno, Isr. J. Chem., 2015, 55, 40–50 CrossRef CAS.
- S. Abe and T. Ueno, RSC Adv., 2015, 5, 21366–21375 RSC.
- S. Howorka, Curr. Opin. Biotechnol., 2011, 22, 485–491 CrossRef CAS PubMed.
- B. Pieters, M. B. van Eldijk, R. J. M. Nolte and J. Mecinovic, Chem. Soc. Rev., 2016, 45, 24–39 RSC.
- S. M. Kuiper, M. Nallani, D. M. Vriezema, J. Cornelissen, J. C. M. van Hest, R. J. M. Nolte and A. E. Rowan, Org. Biomol. Chem., 2008, 6, 4315–4318 CAS.
- R. Peters, I. Louzao and J. C. M. van Hest, Chem. Sci., 2012, 3, 335–342 RSC.
- M. Marguet, C. Bonduelle and S. Lecommandoux, Chem. Soc. Rev., 2013, 42, 512–529 RSC.
- S. F. M. van Dongen, M. Nallani, J. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Chem. – Eur. J., 2009, 15, 1107–1114 CrossRef CAS PubMed.
- M. L. Flenniken, M. Uchida, L. O. Liepold, S. Kang, M. J. Young and T. Douglas, in Viruses and Nanotechnology, ed. M. Manchester and N. F. Steinmetz, 2009, pp. 71–93 Search PubMed.
- T. O. Yeates, M. C. Thompson and T. A. Bobik, Curr. Opin. Struct. Biol., 2011, 21, 223–231 CrossRef CAS PubMed.
- T. O. Yeates, C. A. Kerfeld, S. Heinhorst, G. C. Cannon and J. M. Shively, Nat. Rev. Microbiol., 2008, 6, 681–691 CrossRef CAS PubMed.
- A. de la Escosura, R. J. M. Nolte and J. J. L. M. Cornelissen, J. Mater. Chem., 2009, 19, 2274–2278 RSC.
- K. Renggli, P. Baumann, K. Langowska, O. Onaca, N. Bruns and W. Meier, Adv. Funct. Mater., 2011, 21, 1241–1259 CrossRef CAS.
- Y. Azuma, R. Zschoche, M. Tinzl and D. Hilvert, Angew. Chem., Int. Ed., 2016, 55, 1531–1534 CrossRef CAS PubMed.
- L. Schoonen and J. C. M. van Hest, Nanoscale, 2014, 6, 7124–7141 RSC.
- R. Zschoche and D. Hilvert, J. Am. Chem. Soc., 2015, 137, 16121–16132 CrossRef CAS PubMed.
- M. Comellas-Aragones, H. Engelkamp, V. I. Claessen, N. A. J. M. Sommerdijk, A. E. Rowan, P. C. M. Christianen, J. C. Maan, B. J. M. Verduin, J. J. L. M. Cornelissen and R. J. M. Nolte, Nat. Nanotechnol., 2007, 2, 635–639 CrossRef CAS PubMed.
- D. P. Patterson, B. Schwarz, R. S. Waters, T. Gedeon and T. Douglas, ACS Chem. Biol., 2014, 9, 359–365 CrossRef CAS PubMed.
- H. E. van Kan-Davelaar, J. C. M. van Hest, J. J. L. M. Cornelissen and M. S. T. Koay, Br. J. Pharmacol., 2014, 171, 4001–4009 CrossRef CAS PubMed.
- I. Yildiz, S. Shukla and N. F. Steinmetz, Curr. Opin. Biotechnol., 2011, 22, 901–908 CrossRef CAS PubMed.
- J. Lucon, S. Qazi, M. Uchida, G. J. Bedwell, B. LaFrance, P. E. Prevelige and T. Douglas, Nat. Chem., 2012, 4, 781–788 CrossRef CAS PubMed.
- R. Usselman, S. Qazi, P. Aggarwal, S. Eaton, G. Eaton, S. Russek and T. Douglas, Appl. Magn. Reson., 2015, 46, 349–355 CrossRef CAS.
- M. Brasch, I. K. Voets, M. S. T. Koay and J. J. L. M. Cornelissen, Faraday Discuss., 2013, 166, 47–57 RSC.
- W. Wu, S. C. Hsiao, Z. M. Carrico and M. B. Francis, Angew. Chem., Int. Ed., 2009, 48, 9493–9497 CrossRef CAS PubMed.
- L. Li, C. J. Fang, J. C. Ryan, E. C. Niemi, J. A. Lebron, P. J. Bjorkman, H. Arase, F. M. Torti, S. V. Torti, M. C. Nakamura and W. E. Seaman, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 3505–3510 CrossRef CAS PubMed.
- G. Jutz, P. van Rijn, B. S. Miranda and A. Boker, Chem. Rev., 2015, 115, 1653–1701 CrossRef CAS PubMed.
- S. A. Bode, I. J. Minten, R. J. M. Nolte and J. J. L. M. Cornelissen, Nanoscale, 2011, 3, 2376–2389 RSC.
- X. Liu and E. C. Theil, Acc. Chem. Res., 2005, 38, 167–175 CrossRef CAS PubMed.
- T. Ueno, M. Suzuki, T. Goto, T. Matsumoto, K. Nagayama and Y. Watanabe, Angew. Chem., Int. Ed., 2004, 43, 2527–2530 CrossRef CAS PubMed.
- S. Kanbak-Aksu, M. N. Hasan, W. R. Hagen, F. Hollmann, D. Sordi, R. A. Sheldon and I. Arends, Chem. Commun., 2012, 48, 5745–5747 RSC.
- M. Suzuki, M. Abe, T. Ueno, S. Abe, T. Goto, Y. Toda, T. Akita, Y. Yamadae and Y. Watanabe, Chem. Commun., 2009, 4871–4873 RSC.
- O. D. Petrucci, D. C. Buck, J. K. Farrer and R. K. Watt, RSC Adv., 2014, 4, 3472–3481 RSC.
- C. Sun, H. Yang, Y. Yuan, X. Tian, L. Wang, Y. Guo, L. Xu, J. Lei, N. Gao, G. J. Anderson, X.-J. Liang, C. Chen, Y. Zhao and G. Nie, J. Am. Chem. Soc., 2011, 133, 8617–8624 CrossRef CAS PubMed.
- C. Sun, Y. Yuan, Z. Xu, T. Ji, Y. Tian, S. Wu, J. Lei, J. Li, N. Gao and G. Nie, Bioconjugate Chem., 2015, 26, 193–196 CrossRef CAS PubMed.
- E. Fantechi, C. Innocenti, M. Zanardelli, M. Fittipaldi, E. Falvo, M. Carbo, V. Shullani, L. Di Cesare Mannelli, C. Ghelardini, A. M. Ferretti, A. Ponti, C. Sangregorio and P. Ceci, ACS Nano, 2014, 8, 4705–4719 CrossRef CAS PubMed.
- K. L. Fan, C. Q. Cao, Y. X. Pan, D. Lu, D. L. Yang, J. Feng, L. N. Song, M. M. Liang and X. Y. Yan, Nat. Nanotechnol., 2012, 7, 459–464 CrossRef CAS PubMed.
- C. Cao, X. Wang, Y. Cai, L. Sun, L. Tian, H. Wu, X. He, H. Lei, W. Liu, G. Chen, R. Zhu and Y. Pan, Adv. Mater., 2014, 26, 2566–2571 CrossRef CAS PubMed.
- X. Y. Liu, W. Wei, Q. Yuan, X. Zhang, N. Li, Y. G. Du, G. H. Ma, C. H. Yan and D. Ma, Chem. Commun., 2012, 48, 3155–3157 RSC.
- J. Fan, J.-J. Yin, B. Ning, X. Wu, Y. Hu, M. Ferrari, G. J. Anderson, J. Wei, Y. Zhao and G. Nie, Biomaterials, 2011, 32, 1611–1618 CrossRef CAS PubMed.
- W. Zhang, X. Liu, D. Walsh, S. Yao, Y. Kou and D. Ma, Small, 2012, 8, 2948–2953 CrossRef CAS PubMed.
- X. Liu, W. Wei, C. Wang, H. Yue, D. Ma, C. Zhu, G. Ma and Y. Du, J. Mater. Chem., 2011, 21, 7105–7110 RSC.
- T. D. Bradshaw, M. Junor, A. Patane, P. Clarke, N. R. Thomas, M. Li, S. Mann and L. Turyanska, J. Mater. Chem. B, 2013, 1, 6254–6260 RSC.
- S. Abe, K. Hirata, T. Ueno, K. Morino, N. Shimizu, M. Yamamoto, M. Takata, E. Yashima and Y. Watanabe, J. Am. Chem. Soc., 2009, 131, 6958–6960 CrossRef CAS PubMed.
- S. Abe, J. Niemeyer, M. Abe, Y. Takezawa, T. Ueno, T. Hikage, G. Erker and Y. Watanabe, J. Am. Chem. Soc., 2008, 130, 10512–10514 CrossRef CAS PubMed.
- K. Fujita, Y. Tanaka, T. Sho, S. Ozeki, S. Abe, T. Hikage, T. Kuchimaru, S. Kizaka-Kondoh and T. Ueno, J. Am. Chem. Soc., 2014, 136, 16902–16908 CrossRef CAS PubMed.
- K. Fujita, Y. Tanaka, S. Abe and T. Ueno, Angew. Chem., Int. Ed., 2016, 55, 1056–1060 CrossRef CAS PubMed.
- H. Nakajima, M. Kondo, T. Nakane, S. Abe, T. Nakao, Y. Watanabe and T. Ueno, Chem. Commun., 2015, 51, 16609–16612 RSC.
- Z. Zhen, W. Tang, C. Guo, H. Chen, X. Lin, G. Liu, B. Fei, X. Chen, B. Xu and J. Xie, ACS Nano, 2013, 7, 6988–6996 CrossRef CAS PubMed.
- R. Xing, X. Wang, C. Zhang, Y. Zhang, Q. Wang, Z. Yang and Z. Guo, J. Inorg. Biochem., 2009, 103, 1039–1044 CrossRef CAS PubMed.
- Z. Yang, X. Wang, H. Diao, J. Zhang, H. Li, H. Sun and Z. Guo, Chem. Commun., 2007, 3453–3455 RSC.
- S. Aime, L. Frullano and S. Geninatti Crich, Angew. Chem., Int. Ed., 2002, 41, 1017–1019 CrossRef CAS.
- Y. Song, Y. J. Kang, H. Jung, H. Kim, S. Kang and H. Cho, Sci. Rep., 2015, 5, 15656 CrossRef CAS PubMed.
- S. Geninatti Crich, M. Cadenazzi, S. Lanzardo, L. Conti, R. Ruiu, D. Alberti, F. Cavallo, J. C. Cutrin and S. Aime, Nanoscale, 2015, 7, 6527–6533 RSC.
- J. C. Cutrin, S. G. Crich, D. Burghelea, W. Dastrù and S. Aime, Mol. Pharmaceutics, 2013, 10, 2079–2085 CrossRef CAS PubMed.
- I. Szabo, S. G. Crich, D. Alberti, F. K. Kalman and S. Aime, Chem. Commun., 2012, 48, 2436–2438 RSC.
- B. Sana, C. L. Poh and S. Lim, Chem. Commun., 2012, 48, 862–864 RSC.
- X. Lin, J. Xie, G. Niu, F. Zhang, H. Gao, M. Yang, Q. Quan, M. A. Aronova, G. Zhang, S. Lee, R. Leapman and X. Chen, Nano Lett., 2011, 11, 814–819 CrossRef CAS PubMed.
- X. Liu, Z. Ye, W. Wei, Y. Du, J. Yuan and D. Ma, Chem. Commun., 2011, 47, 8139–8141 RSC.
- L. Tian, Z. Dai, X. Liu, B. Song, Z. Ye and J. Yuan, Anal. Chem., 2015, 87, 10878–10885 CrossRef CAS PubMed.
- M. M. Liang, K. L. Fan, M. Zhou, D. M. Duan, J. Y. Zheng, D. L. Yang, J. Feng and X. Y. Yan, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14900–14905 CrossRef CAS PubMed.
- Z. Zhen, W. Tang, H. Chen, X. Lin, T. Todd, G. Wang, T. Cowger, X. Chen and J. Xie, ACS Nano, 2013, 7, 4830–4837 CrossRef CAS PubMed.
- M. L. Flenniken, L. O. Liepold, B. E. Crowley, D. A. Willits, M. J. Young and T. Douglas, Chem. Commun., 2005, 447–449 RSC.
- F. Yan, Y. Zhang, H.-k. Yuan, M. K. Gregas and T. Vo-Dinh, Chem. Commun., 2008, 4579–4581 RSC.
- P. Huang, P. Rong, A. Jin, X. Yan, M. G. Zhang, J. Lin, H. Hu, Z. Wang, X. Yue, W. Li, G. Niu, W. Zeng, W. Wang, K. Zhou and X. Chen, Adv. Mater., 2014, 26, 6401–6408 CrossRef CAS PubMed.
- N. G. Belén Fernández, P. Sánchez, R. Cuesta, R. Bermejo and J. M. Domínguez-Vera, JBIC, J. Biol. Inorg. Chem., 2008, 13, 349–355 CrossRef PubMed.
- K. Fan, C. Cao, Y. Pan, D. Lu, D. Yang, J. Feng, L. Song, M. Liang and X. Yan, Nat. Nanotechnol., 2012, 7, 459–464 CrossRef CAS PubMed.
- T. Ueno, M. Abe, K. Hirata, S. Abe, M. Suzuki, N. Shimizu, M. Yamamoto, M. Takata and Y. Watanabe, J. Am. Chem. Soc., 2009, 131, 5094–5100 CrossRef CAS PubMed.
- S. Abe, T. Ueno and Y. Watanabe, in Bio-Inspired Catalysts, ed. T. R. Ward, 2009, pp. 25–43 Search PubMed.
- W. Arap, R. Pasqualini and E. Ruoslahti, Science, 1998, 279, 377–380 CrossRef CAS PubMed.
- Z. Zhen, W. Tang, W. Zhang and J. Xie, Nanoscale, 2015, 7, 10330–10333 RSC.
- R. Motterlini and L. E. Otterbein, Nat. Rev. Drug Discovery, 2010, 9, 728–743 CrossRef CAS PubMed.
- W. Cai, C.-C. Chu, G. Liu and Y.-X. J. Wáng, Small, 2015, 11, 4806–4822 CrossRef CAS PubMed.
- S. Keskin and S. Kızılel, Ind. Eng. Chem. Res., 2011, 50, 1799–1812 CrossRef CAS.
- C. G. Palivan, R. Goers, A. Najer, X. Zhang, A. Car and W. Meier, Chem. Soc. Rev., 2016, 45, 377–411 RSC.
- P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J.-S. Chang, Y. K. Hwang, V. Marsaud, P.-N. Bories, L. Cynober, S. Gil, G. Ferey, P. Couvreur and R. Gref, Nat. Mater., 2010, 9, 172–178 CrossRef CAS PubMed.
- K. Langowska, C. G. Palivan and W. Meier, Chem. Commun., 2013, 49, 128–130 RSC.
- J. C. Kendrew, G. Bodo, H. M. Dintzis, R. G. Parrish, H. Wyckoff and D. C. Phillips, Nature, 1958, 181, 662–666 CrossRef CAS PubMed.
- A. L. Margolin and M. A. Navia, Angew. Chem., Int. Ed., 2001, 40, 2205–2222 CrossRef.
- M. Guli, E. M. Lambert, M. Li and S. Mann, Angew. Chem., Int. Ed., 2010, 49, 520–523 CrossRef CAS PubMed.
- H. Wei, Z. D. Wang, J. O. Zhang, S. House, Y. G. Gao, L. M. Yang, H. Robinson, L. H. Tan, H. Xing, C. J. Hou, I. M. Robertson, J. M. Zuo and Y. Lu, Nat. Nanotechnol., 2011, 6, 92–96 CrossRef PubMed.
- H. Wei and Y. Lu, Chem. – Asian J., 2012, 7, 680–683 CrossRef CAS PubMed.
- M. Liang, L. B. Wang, X. Liu, W. Qi, R. X. Su, R. L. Huang, Y. J. Yu and Z. M. He, Nanotechnology, 2013, 24, 245601 CrossRef PubMed.
- M. Liang, L. B. Wang, R. X. Su, W. Qi, M. F. Wang, Y. J. Yu and Z. M. He, Catal. Sci. Technol., 2013, 3, 1910–1914 CAS.
- S. Abe, M. Tsujimoto, K. Yoneda, M. Ohba, T. Hikage, M. Takano, S. Kitagawa and T. Ueno, Small, 2012, 8, 1314–1319 CrossRef CAS PubMed.
- M. W. England, E. M. Lambert, M. Li, L. Turyanska, A. J. Patil and S. Mann, Nanoscale, 2012, 4, 6710–6713 RSC.
- M. W. England, A. J. Patil and S. Mann, Chem. – Eur. J., 2015, 21, 9008–9013 CrossRef CAS PubMed.
- H. Wei, S. House, J. J. X. Wu, J. Zhang, Z. D. Wang, Y. He, E. J. Gao, Y. G. Gao, H. Robinson, W. Li, J. M. Zuo, I. M. Robertson and Y. Lu, Nano Res., 2013, 6, 627–634 CrossRef CAS.
- T. Ueno, S. Abe, T. Koshiyama, T. Ohki, T. Hikage and Y. Watanabe, Chem. – Eur. J., 2010, 16, 2730–2740 CrossRef CAS PubMed.
- H. Tabe, S. Abe, T. Hikage, S. Kitagawa and T. Ueno, Chem. – Asian J., 2014, 9, 1373–1378 CrossRef CAS PubMed.
- H. Tabe, K. Fujita, S. Abe, M. Tsujimoto, T. Kuchimaru, S. Kizaka-Kondoh, M. Takano, S. Kitagawa and T. Ueno, Inorg. Chem., 2015, 54, 215–220 CrossRef CAS PubMed.
- T. Koshiyama, M. Shirai, T. Hikage, H. Tabe, K. Tanaka, S. Kitagawa and T. Ueno, Angew. Chem., Int. Ed., 2011, 50, 4849–4852 CrossRef CAS PubMed.
- T. Koshiyama, N. Kawaba, T. Hikage, M. Shirai, Y. Miura, C. Y. Huang, K. Tanaka, Y. Watanabe and T. Ueno, Bioconjugate Chem., 2010, 21, 264–269 CrossRef CAS PubMed.
- P. A. Sontz, J. B. Bailey, S. Aln and F. A. Tezcan, J. Am. Chem. Soc., 2015, 137, 11598–11601 CrossRef CAS PubMed.
- V. Liljestrom, J. Seitsonen and M. A. Kostiainen, ACS Nano, 2015, 9, 11278–11285 CrossRef CAS PubMed.
- J. Mikkila, E. Anaya-Plaza, V. Liljestrom, J. R. Caston, T. Torres, A. Escosura and M. A. Kostiainen, ACS Nano, 2016, 10, 1565–1571 CrossRef CAS PubMed.
- Y. T. Lai, E. Reading, G. L. Hura, K. L. Tsai, A. Laganowsky, F. J. Asturias, J. A. Tainer, C. V. Robinson and T. O. Yeates, Nat. Chem., 2014, 6, 1065–1071 CrossRef CAS PubMed.
- V. Liljestrom, J. Mikkila and M. A. Kostiainen, Nat. Commun., 2014, 5, 4445 CAS.
- G. Cattani, L. Vogeley and P. B. Crowley, Nat. Chem., 2015, 7, 823–828 CrossRef CAS PubMed.
- L. Z. Vilenchik, J. P. Griffith, N. St Clair, M. A. Navia and A. L. Margolin, J. Am. Chem. Soc., 1998, 120, 4290–4294 CrossRef CAS.
- J. J. Lalonde, C. Govardhan, N. Khalaf, A. G. Martinez, K. Visuri and A. L. Margolin, J. Am. Chem. Soc., 1995, 117, 6845–6852 CrossRef CAS.
- Y. B. Ding, L. L. Shi and H. Wei, J. Mater. Chem. B, 2014, 2, 8268–8291 RSC.
- M. Razavet, V. Artero, C. Cavazza, Y. Oudart, C. Lebrun, J. C. Fontecilla-Camps and M. Fontecave, Chem. Commun., 2007, 2805–2807 RSC.
- I. W. McNae, K. Fishburne, A. Habtemariam, T. M. Hunter, M. Melchart, F. Y. Wang, M. D. Walkinshaw and P. J. Sadler, Chem. Commun., 2004, 1786–1787 RSC.
- H. Oki, Y. Matsuura, H. Komatsu and A. A. Chernov, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1999, 55, 114–121 CrossRef CAS PubMed.
- M. C. Vaney, S. Maignan, M. RiesKautt and A. Ducruix, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1996, 52, 505–517 CrossRef CAS PubMed.
- S. T. Rao and M. Sundaralingam, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1996, 52, 170–175 CrossRef CAS PubMed.
- P. A. Sontz, W. J. Song and F. A. Tezcan, Curr. Opin. Chem. Biol., 2014, 19, 42–49 CrossRef CAS PubMed.
- D. J. E. Huard, K. M. Kane and F. A. Tezcan, Nat. Chem. Biol., 2013, 9, 169–176 CrossRef CAS PubMed.
- J. P. K. Doye and W. C. K. Poon, Curr. Opin. Colloid Interface Sci., 2006, 11, 40–46 CrossRef CAS.
- H. Hasegawa, J. Wendling, F. He, E. Trilisky, R. Stevenson, H. Franey, F. Kinderman, G. Li, D. M. Piedmonte, T. Osslund, M. Shen and R. R. Ketchem, J. Biol. Chem., 2011, 286, 19917–19931 CrossRef CAS PubMed.
- L. Redecke, K. Nass, D. P. DePonte, T. A. White, D. Rehders, A. Barty, F. Stellato, M. N. Liang, T. R. M. Barends, S. Boutet, G. J. Williams, M. Messerschmidt, M. M. Seibert, A. Aquila, D. Arnlund, S. Bajt, T. Barth, M. J. Bogan, C. Caleman, T. C. Chao, R. B. Doak, H. Fleckenstein, M. Frank, R. Fromme, L. Galli, I. Grotjohann, M. S. Hunter, L. C. Johansson, S. Kassemeyer, G. Katona, R. A. Kirian, R. Koopmann, C. Kupitz, L. Lomb, A. V. Martin, S. Mogk, R. Neutze, R. L. Shoeman, J. Steinbrener, N. Timneanu, D. J. Wang, U. Weierstall, N. A. Zatsepin, J. C. H. Spence, P. Fromme, I. Schlichting, M. Duszenko, C. Betzel and H. N. Chapman, Science, 2013, 339, 227–230 CrossRef CAS PubMed.
- M. Duszenko, L. Redecke, C. N. Mudogo, B. P. Sommer, S. Mogk, D. Oberthuer and C. Betzel, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2015, 71, 929–937 CrossRef CAS PubMed.
- F. Coulibaly, E. Chiu, K. Ikeda, S. Gutmann, P. Haebel, C. Schulze-Briese, H. Mori and P. Metcalf, Nature, 2007, 446, 97–101 CrossRef CAS PubMed.
- R. Koopmann, K. Cupelli, L. Redecke, K. Nass, D. P. DePonte, T. A. White, F. Stellato, D. Rehders, M. N. Liang, J. Andreasson, A. Aquila, S. Bajt, M. Barthelmess, A. Barty, M. J. Bogan, C. Bostedt, S. Boutet, J. D. Bozek, C. Caleman, N. Coppola, J. Davidsson, R. B. Doak, T. Ekeberg, S. W. Epp, B. Erk, H. Fleckenstein, L. Foucar, H. Graafsma, L. Gumprecht, J. Hajdu, C. Y. Hampton, A. Hartmann, R. Hartmann, G. Hauser, H. Hirsemann, P. Holl, M. S. Hunter, S. Kassemeyer, R. A. Kirian, L. Lomb, F. Maia, N. Kimmel, A. V. Martin, M. Messerschmidt, C. Reich, D. Rolles, B. Rudek, A. Rudenko, I. Schlichting, J. Schulz, M. M. Seibert, R. L. Shoeman, R. G. Sierra, H. Soltau, S. Stern, L. Struder, N. Timneanu, J. Ullrich, X. Y. Wang, G. Weidenspointner, U. Weierstall, G. J. Williams, C. B. Wunderer, P. Fromme, J. C. H. Spence, T. Stehle, H. N. Chapman, C. Betzel and M. Duszenko, Nat. Methods, 2012, 9, 259–262 CrossRef CAS PubMed.
- M. R. Sawaya, D. Cascio, M. Gingery, J. Rodriguez, L. Goldschmidt, J. P. Colletier, M. M. Messerschmidt, S. Boutet, J. E. Koglin, G. J. Williams, A. S. Brewster, K. Nass, J. Hattne, S. Botha, R. B. Doak, R. L. Shoeman, D. P. DePonte, H. W. Park, B. A. Federici, N. K. Sauter, I. Schlichting and D. S. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 12769–12774 CrossRef CAS PubMed.
- E. Chiu, M. Hijnen, R. D. Bunker, M. Boudes, C. Rajendran, K. Aizel, V. Olieric, C. Schulze-Briese, W. Mitsuhashi, V. Young, V. K. Ward, M. Bergoin, P. Metcalf and F. Coulibaly, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 3973–3978 CrossRef CAS PubMed.
- H. Tsutsui, Y. Jinno, K. Shoda, A. Tomita, M. Matsuda, E. Yamashita, H. Katayama, A. Nakagawa and A. Miyawaki, Mol. Cell, 2015, 58, 186–193 CrossRef CAS PubMed.
- F. Coulibaly, E. Chiu, S. Gutmann, C. Rajendran, P. W. Haebel, K. Ikeda, H. Mori, V. K. Ward, C. Schulze-Briese and P. Metcalf, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 22205–22210 CrossRef CAS PubMed.
- D. H. Kim, H. Lee, Y. K. Lee, J. M. Nam and A. Levchenko, Adv. Mater., 2010, 22, 4551–4566 CrossRef CAS PubMed.
- D. Axford, X. Y. Ji, D. I. Stuart and G. Sutton, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2014, 70, 1435–1441 CrossRef CAS PubMed.
- H. Ijiri, F. Coulibaly, G. Nishimura, D. Nakai, E. Chiu, C. Takenaka, K. Ikeda, H. Nakazawa, N. Hamada, E. Kotani, P. Metcalf, S. Kawamata and H. Mori, Biomaterials, 2009, 30, 4297–4308 CrossRef CAS PubMed.
- H. Mori, C. Shukunami, A. Furuyama, H. Notsu, Y. Nishizaki and Y. Hiraki, J. Biol. Chem., 2007, 282, 17289–17296 CrossRef CAS PubMed.
- N. Nishishita, H. Ijiri, C. Takenaka, K. Kobayashi, K. Goto, E. Kotani, T. Itoh, H. Mori and S. Kawamata, Biomaterials, 2011, 32, 3555–3563 CrossRef CAS PubMed.
- G. Matsumoto, R. Hirohata, K. Hayashi, Y. Sugimoto, E. Kotani, J. Shimabukuro, T. Hirano, Y. Nakajima, S. Kawamata and H. Mori, Biomaterials, 2014, 35, 1326–1333 CrossRef CAS PubMed.
- S. Abe, H. Ijiri, H. Negishi, H. Yamanaka, K. Sasaki, K. Hirata, H. Mori and T. Ueno, Adv. Mater., 2015, 27, 7951–7956 CrossRef CAS PubMed.
- S. Abe, Y. Tokura, R. Pal, N. Komura, A. Imamura, K. Matsumoto, H. Ijiri, N. J. M. Sanghamitra, H. Tabe, H. Ando, M. Kiso, H. Mori, S. Kitagawa and T. Ueno, Chem. Lett., 2015, 44, 29–31 CrossRef CAS.
- H. Tabe, T. Shimoi, K. Fujita, S. Abe, H. Ijiri, M. Tsujimoto, T. Kuchimaru, S. Kizaka-Kondo, H. Mori, S. Kitagawa and T. Ueno, Chem. Lett., 2015, 44, 342–344 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |