
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
[18]Annulene put into a new perspective†‡
Dominik
Lungerich
a,
Alexey V.
Nizovtsev
b,
Frank W.
Heinemann
b,
Frank
Hampel
a,
Karsten
Meyer
b,
George
Majetich
c,
Paul v. R.
Schleyer
c and
Norbert
Jux
*a
aDepartment Chemie und Pharmazie & Interdisciplinary Center for Molecular Materials (ICMM), Organic Chemistry II, Friedrich-Alexander-Universität Erlangen–Nürnberg, Henkestraße 42, 91054, Erlangen, Germany. E-mail: norbert.jux@fau.de
bDepartment Chemie und Pharmazie, Inorganic Chemistry, Friedrich-Alexander-Universität Erlangen-Nürnberg, Egerlandstraße 1, 91058, Erlangen, Germany
cDepartment of Chemistry, University of Georgia, 140 Cedar St, Athens, GA 30602, USA
First published on 2nd March 2016
Abstract
New insights into [18]annulene were gained by looking more closely at its X-ray structure, revealing a close face-to-face stacking of 3.16 Å in a herringbone-like crystal packing. Hexadehydro[18]annulene was co-crystalized in a benzene matrix, demonstrating the stabilizing role of intercalated solvent molecules in solid annulenes.
The story of [18]annulene 1 – its structural elucidation and its aromaticity – is probably one of the most controversially discussed issues in the field of physical organic chemistry, even though it represents one of the classic molecules found in virtually every organic and spectroscopy textbook.1,2 Originally, with its 18π electrons, its preparation served the understanding of larger aromatic compounds3,4 that follow Hückels (4n + 2)π rule and the distinction, up to which ring size aromaticity has an effect on bond length alternation and a stabilizing effect on the molecule.5–7 Down to the present day, 1 has set off an avalanche of publications mostly theoretical in nature concerning e.g., the re-evaluation of its aromaticity,8 or as a model compound for the ring-current in porphyrin 2 and related tetrapyrroles.9 However, as for experimental research, 1 has withered away from the current literature, most likely due to its tendency to decompose, accompanied by unattractive overall yields of 0.4 to 0.6%, respectively.10,11
Accompanied by a modern and improved synthesis, we discuss here [18]annulene 1 for the first time from a materials chemical perspective.12 We re-investigated its brown crystals, grown from diethyl ether and methanol at −20 °C, which were elucidated by means of X-ray diffraction. 1 crystalizes in the space group P21/n as shown in Fig. 1c. We found essentially the same structural characteristics as already described in 1995.4 Taking a closer look at the larger crystal packing, presented in Fig. 1a and b, we were exhilarated by its similarity to the herringbone packing of pentacene 3a.13 However, a critical difference between 3a and 1 is given by the very intimate face-to-face distance of only 3.16 Å between the respective π-surfaces (see Fig. 2a and c) in 1. Comparing the distances between individual graphene sheets in graphite – 3.35 Å,14 the layers in crystals of TIPS-pentacene 3b – 3.43 Å,15 or in coronene 4 – 3.46 Å,16 the reduced face-to-face association of up to 0.4 Å in 1 is significant. The discrepancy between layers of 1 and 4 – which share the same outer carbon skeleton – can be explained by the “annulene-hole”. The van der Waals radii of the inner C–H bonds in 1 are around 0.4 Å smaller in size than those of the respective C–C bonds in 4, resulting in closer π–π stacks. Thus, the repulsive vdW forces of the outer carbon framework of 1 remain essentially the same as in 4. However, the slipped stacking of 1 that allows overlap in the “annulene-hole” region, which is non-existent in 4, is most likely responsible for this closer interaction.4 Hence, directly overlapping stacks of 1 would probably result in comparable stacking distances as in coronene 4. The exact nature of this effect remains not fully understood yet.§Fig. 2b shows the HOMO and the LUMO of 1. Both MOs are distributed equally over the cyclic molecule, assuming a sufficient electronic communication in the solid state. The C–C edge-to-face distance is 3.52 Å and the molecules stand at an angle of 82.4° to each other, respectively. We investigated thin-films of 1 by means of solid state absorption spectroscopy. Therefore, we drop-cast 1 from a 10−5 M CH2Cl2 solution, which was evaporated under a continuous N2-flow. As shown in Fig. 3, 1 shows distinct absorption maxima at 375 nm (300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1) and at 454 nm (25000 M−1 cm−1) in solution. The optical bandgap Eg was estimated from solution (CH2Cl2), based on the intercept of a tangent applied to the lower edge of the longest wavelength absorption and the x-axis, indicating an energy gap of 2.64 eV (469 nm). The solid state spectrum of 1 (dashed line in Fig. 3) shows broadened absorption characteristics and a peculiar red-shift of 7 and 6 nm to 382 and 460 nm, respectively. The bathochromic shift indicates close π-stacks, similar to those depicted in the crystal packing. Even though there is still much controversy regarding the electronic transport in organic materials, it remains irrefutable that interlocked layers in herringbone-like structured van der Waals crystals are prone to higher field-effect mobilities.17 Nonetheless, we relinquished to perform actual device experiments with 1, since it is clearly not a realistic candidate. Its thermal instability and its tendency to decompose rapidly are probably the most significant arguments not to study its device applicability.17 However, functionalized non-benzenoid derivatives of 1 that make use of the “annulene-hole” in order to get into close face-to-face contact, might become an important strategy for the design of organic materials.
000 M−1 cm−1) and at 454 nm (25000 M−1 cm−1) in solution. The optical bandgap Eg was estimated from solution (CH2Cl2), based on the intercept of a tangent applied to the lower edge of the longest wavelength absorption and the x-axis, indicating an energy gap of 2.64 eV (469 nm). The solid state spectrum of 1 (dashed line in Fig. 3) shows broadened absorption characteristics and a peculiar red-shift of 7 and 6 nm to 382 and 460 nm, respectively. The bathochromic shift indicates close π-stacks, similar to those depicted in the crystal packing. Even though there is still much controversy regarding the electronic transport in organic materials, it remains irrefutable that interlocked layers in herringbone-like structured van der Waals crystals are prone to higher field-effect mobilities.17 Nonetheless, we relinquished to perform actual device experiments with 1, since it is clearly not a realistic candidate. Its thermal instability and its tendency to decompose rapidly are probably the most significant arguments not to study its device applicability.17 However, functionalized non-benzenoid derivatives of 1 that make use of the “annulene-hole” in order to get into close face-to-face contact, might become an important strategy for the design of organic materials.
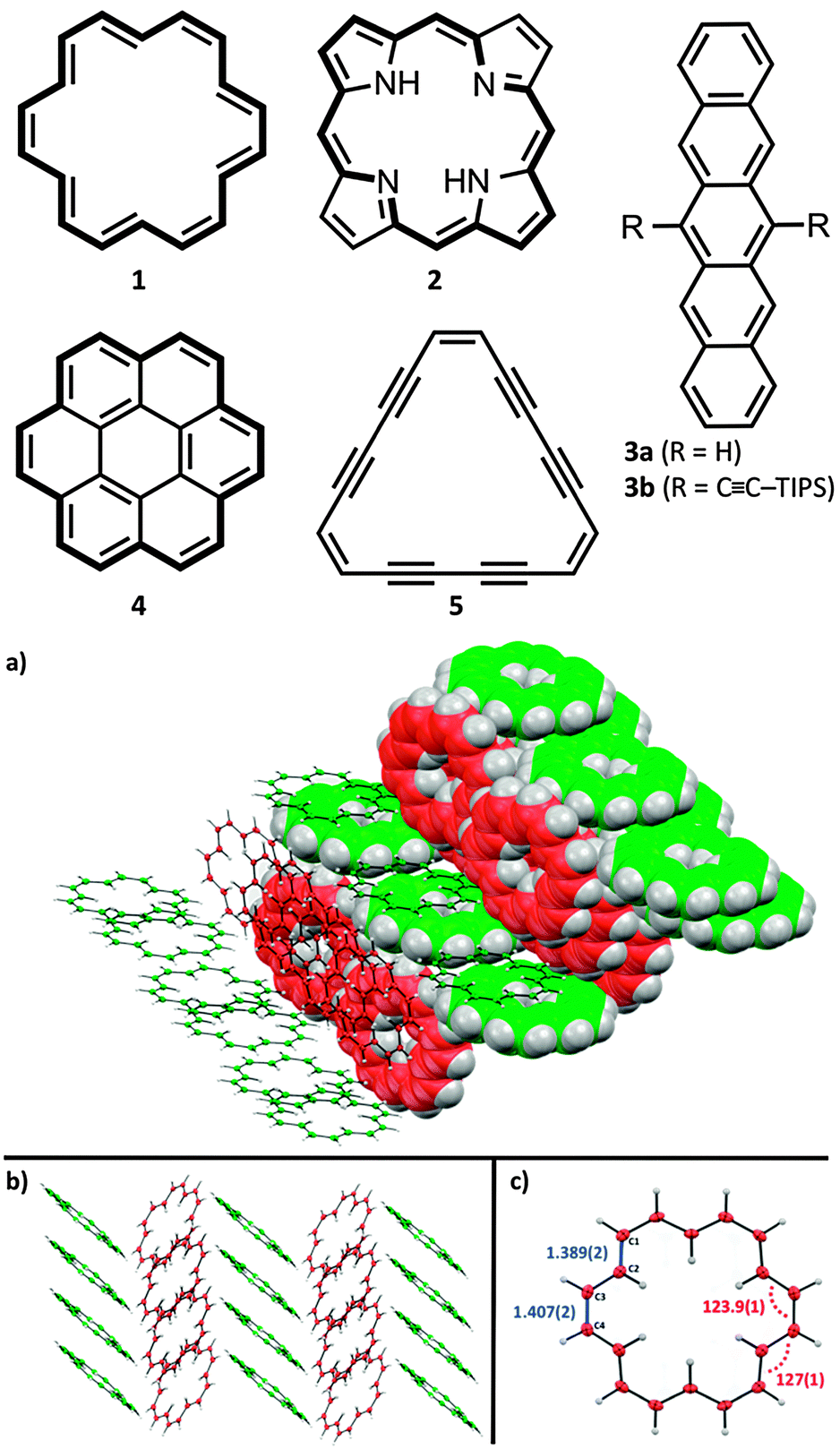 | ||
| Fig. 1 X-ray structure analysis of 1; (a) crystal packing depicted as space-filling model and as ORTEP model; (b) side view of herringbone pattern; (c) crystal structure, showing averaged bond lengths indicated in blue and averaged bond angles depicted in red; ORTEP models are depicted as 50% thermal ellipsoids. | ||
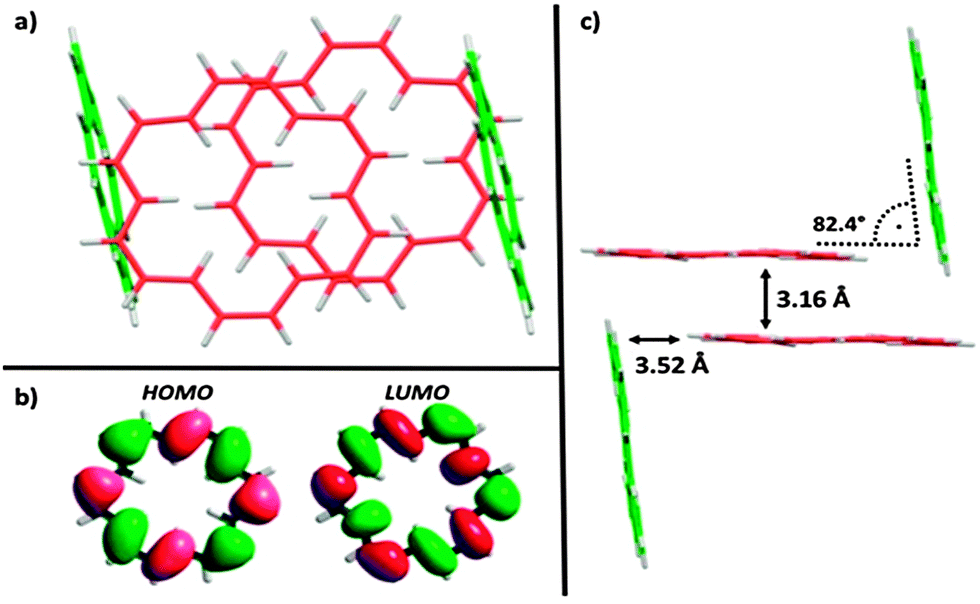 | ||
| Fig. 2 Analysis of 1: (a) top-view of molecular overlap in crystal packing; (b) DFT calculation of HOMO and LUMO at the B3LYP/6-311G* level of theory;¶ (c) side-view of crystal packing. | ||
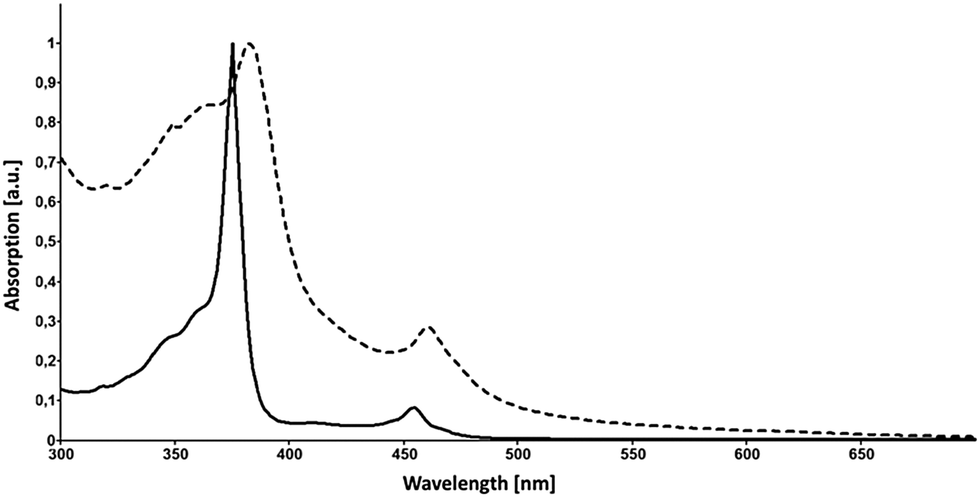 | ||
| Fig. 3 Absorption spectra of 1 in solution (CH2Cl2, solid line) and in the solid state (dashed line). | ||
A pivotal role in the synthesis of 1 is played by the triangular hexadehydro[18]annulene 5. Even though it has been reported nearly 50 years ago,18,19 we were quite surprised not to find any discussion about its crystal structure and crystal packing in the literature.20 This might be due to its well-known instability in the solid state and even explosive decomposition above 85 °C.18,20 We grew stable amber single crystals of 5 from a slowly evaporated mixture of benzene and diethyl ether at −20 °C. Although 5 represents an 18π aromatic compound, the structure shows clearly bond length alternation of localized single-, double-, and triple bonds.21 Bond lengths and – angles are shown in Fig. 4a. 5 crystallizes in the tetragonal space group I41/acd and adapts an almost planar geometry. It shows a parallel arranged packing, in which benzene molecules are co-crystalized in an array of small nano-channels, formed by 5 (compare Fig. 4b–d). Unlike in crystals of 1, the layer distance is increased to 3.60 Å and consequently exceeds even the values reached by 3b and 4. This range jump is most likely explained by the intercalated benzene molecules that serve as a barrier for structural decay. The most likely decomposition mechanism in dehydroannulenes represents the [2+2] alkyne–alkyne polymerization.2,22 However, the symmetrically arranged molecules in the co-crystalized benzene framework are rather badly positioned for [2+2] polymerizations. Nonetheless, under freeze-drying conditions – the slow removal of benzene at low temperatures – 5 shows a rapid decomposition to black amorphous carbon within a few hours, underlining the importance of intercalated solvent molecules in highly unsaturated hydrocarbons.
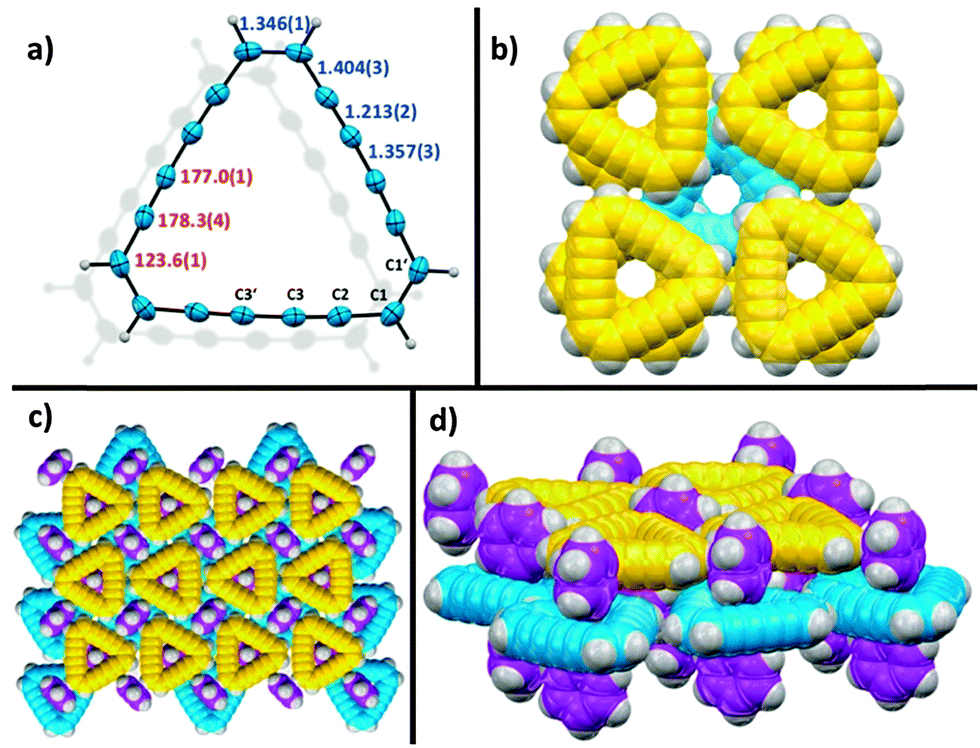 | ||
| Fig. 4 X-ray structure analysis of 5; (a) crystal structure, showing averaged bond lengths indicated in blue and averaged bond angles depicted in red; ORTEP models are depicted as 50% thermal ellipsoids (b) top-view on crystal packing depicted as space-filling model, showing the nano-channel array; benzene molecules are omitted for clarity; (c) top-view on crystal packing depicted as space-filling model; (d) perspective side-view on crystal packing; benzene molecules are depicted in purple for clarity. | ||
1 and 5 were synthesized by a revisited and feasible protocol that takes advantage of modern laboratorial techniques (Scheme 1). We coupled commercially available cis-1,2-dichloroethene and TMS-acetylene with 4% Pd(PPh3)2Cl2 and 2% CuI in n-butylamine and benzene and obtained 6 in 90% yield.236 was deprotected with tetrabutylammonium fluoride (TBAF) in THF at 0 °C, followed by a subsequent oxidative cyclization under modified Eglinton conditions, indicating excess of copper(II) acetate in pyridine at room temperature.24 At an estimated molarity of 45 mM solution of hexenediyne 7 in the pyridine/Cu mixture, yields of up to 24% of 5 were obtained. As reported earlier, the selective reduction of 5 to 1 remains difficult, since [18]annulene 1 is hydrogenated faster than hexadehydro[18]annulene 5.3,10 Hydrogenation of 5 with 5% Pd/CaCO3/Pb (Lindlar catalyst) that was additionally poisoned with quinoline under 1 atm H2 gas was terminated upon complete consumption of the starting material (typically after one hour). 1 was obtained in an average yield of 20%. The overall yield of 4% over three steps remains low; however, it represents the highest reported yield of 1.10,11 Lastly, regarding the stability of 1 and 5, we found a very convenient way to store these annulenes, without any observed decomposition for several months. Therefore, a dilute frozen solution of 1 or 5 in benzene at −20 °C served very well as a protecting matrix.
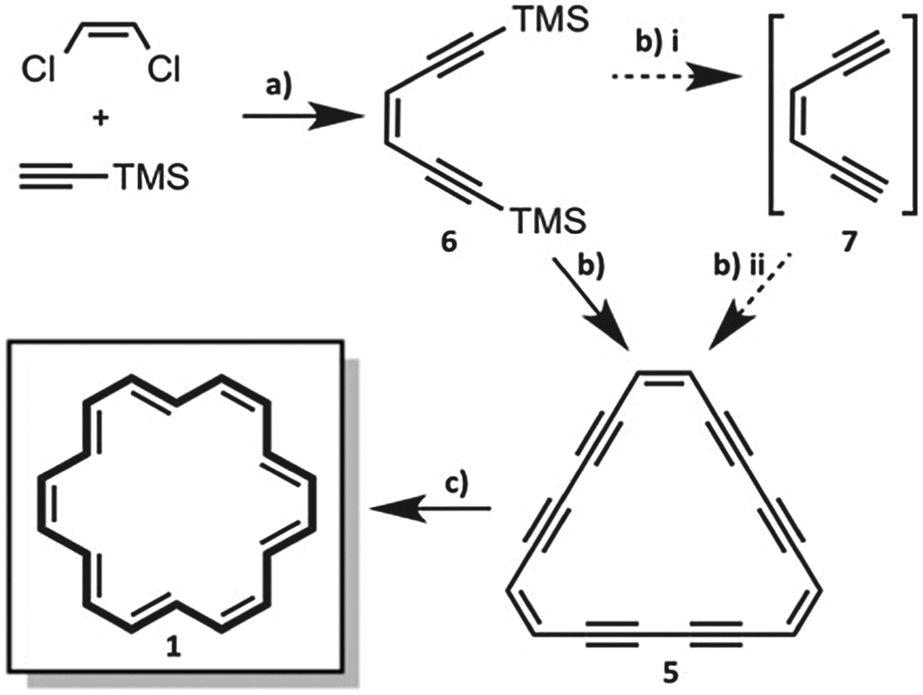 | ||
| Scheme 1 Improved synthesis of 1 and 5; (a) 2% Pd(PPh3)2Cl2, 4% CuI in benzene/n-butylamine, r.t. (90%); (b) (i) 1 M TBAF in THF, 0 °C, (ii) excess Cu(OAc)2·H2O in pyridine, r.t. (24%); (c) 5% Pd/CaCO3/Pb, quinoline, 1 atm H2 in benzene, r.t. (20%). | ||
To summarize, we improved the synthesis of [18]annulene 1 in a three-step protocol with yields exceeding reported values by a factor of ten. With the first X-ray structure of hexadehydro[18]annulene 5, co-crystalized in a benzene matrix, we were able to discuss the role of intercalated solvent residues as a protector towards decomposition to amorphous carbon. The crystal structure analysis of 1 revealed a herringbone-like stacking pattern, with face-to-face distances as low as 3.16 Å, thus undercutting the π–π-contacts of pentacene and coronene by a value of 0.4 Å. This distance discrepancy is attributed to the inner C–H bonds, the “annulene-hole”. By solid state UV/vis experiments, we observed a red-shift of the absorption characteristics that are most likely caused by stacking phenomena. We believe that engineering the periphery of annulenes (excluding benzannelation) will lead to unique properties, concerning material chemical performances and might establish a new class in organic electronics. Derivatization of 1 and device experiments are currently under investigation.
Notes and references
- F. Sondheimer and R. Wolovsky, Tetrahedron Lett., 1959, 1, 3–6 CrossRef.
- E. L. Spitler, C. A. Johnson and M. M. Haley, Chem. Rev., 2006, 106, 5344–5386 CrossRef CAS PubMed.
- F. Sondheimer, R. Wolovsky and Y. Amiel, J. Am. Chem. Soc., 1962, 84, 274–284 CrossRef.
- S. Gorter, E. Rutten-Keulemans, M. Krever, C. Romers and D. W. J. Cruickshank, Acta Crystallogr., Sect. B: Struct. Sci., 1995, 51, 1036–1045 CrossRef.
- J. F. M. Oth, J.-C. Bünzli and Y. de J. Zélicourt, Helv. Chim. Acta, 1974, 57, 2276–2288 CrossRef CAS.
- K. Stöckel, P. J. Garratt and F. Sondheimer, J. Am. Chem. Soc., 1972, 94, 8644–8645 CrossRef.
- S. C. A. H. Pierrefixe and F. M. Bickelhaupt, J. Phys. Chem. A, 2008, 112, 12816–12822 CrossRef CAS PubMed.
- C. S. Wannere, K. W. Sattelmeyer, H. F. Schaefer and P. V. R. Schleyer, Angew. Chem., Int. Ed., 2004, 43, 4200–4206 CrossRef CAS PubMed; C. S. Wannere and P. V. R. Schleyer, Org. Lett., 2003, 5, 865–868 CrossRef PubMed; C. H. Choi, M. Kertesz and A. Karpfen, J. Am. Chem. Soc., 1997, 119, 11994–11995 CrossRef; K. Jug and E. Fasold, J. Am. Chem. Soc., 1987, 109, 2263–2265 CrossRef.
- T. D. Lash, A. Sun, T. Chaney and D. T. Richter, J. Org. Chem., 1998, 63, 9076–9088 CrossRef CAS; T. D. Lash, S. A. Jones and G. M. Ferrence, J. Am. Chem. Soc., 2010, 132, 12786–12787 CrossRef PubMed; H. Fliegl and D. Sundholm, J. Org. Chem., 2012, 77, 3408–3414 CrossRef PubMed; J. I. Wu, I. Fernández and P. V. R. Schleyer, J. Am. Chem. Soc., 2013, 135, 315–321 CrossRef PubMed; M. K. Cyrañski, T. M. Krygowski, M. Wisiorowski, N. J. R. van Eikema Hommes and P. V. R. Schleyer, Angew. Chem., Int. Ed., 1998, 37, 177–180 CrossRef.
- H. P. Figeys and M. Gelbcke, Tetrahedron Lett., 1970, 5139–5142 CrossRef CAS.
- K. Stöckel and F. Sondheimer, Org. Synth., 1974, 54, 1 CrossRef.
- M. Iyoda, J. Yamakawa and M. J. Rahman, Angew. Chem., Int. Ed., 2011, 50, 10522–10553 CrossRef CAS PubMed.
- C. C. Mattheus, A. B. Dros, J. Baas, A. Meetsma, J. L. de Boer and T. T. Palstra, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2001, 57, 939–941 CAS.
- N. Wiberg, Lehrbuch der Anorganischen Chemie, Walter de Gruyter & Co., 2007, pp. 864–865 Search PubMed.
- J. E. Anthony, J. S. Brooks, D. L. Eaton and S. R. Parkin, J. Am. Chem. Soc., 2001, 123, 9482–9483 CrossRef CAS PubMed.
- J. Monteath Robertson and J. G. White, J. Chem. Soc., 1945, 607–617 RSC.
- H. Klauk, Organic electronics, Materials, Manufacturing, and Applications, Wiley-VCH Verlag GmbH, 2006, pp. 39–41 Search PubMed.
- W. H. Okamura and F. Sondheimer, J. Am. Chem. Soc., 1967, 89, 5991–5992 CrossRef CAS.
- F. Sondheimer, Y. Amiel and Y. Gaoni, J. Am. Chem. Soc., 1962, 84, 270–274 CrossRef CAS.
- M. Suzuki, A. Comito, S. I. Khan and Y. Rubin, Org. Lett., 2010, 12, 2346–2349 CrossRef CAS PubMed.
- J. Juselius and D. Sundholm, Phys. Chem. Chem. Phys., 2001, 3, 2433–2437 RSC.
- Q. Zhou, P. J. Carroll and T. M. Swager, J. Org. Chem., 1994, 59, 1294–1301 CrossRef CAS; M. Laskoski, W. Steffen, J. G. M. Morton, M. D. Smith and U. H. F. Bunz, J. Am. Chem. Soc., 2002, 124, 13814–13818 CrossRef PubMed.
- D. Chemin and G. Linstrumelle, Tetrahedron, 1994, 50, 5335–5344 CrossRef CAS.
- G. Eglinton and A. R. Galbraith, Chemistry and Industry, 1956, p. 737 CrossRef CAS; F. Sondheimer, Y. Amiel and R. Wolovsky, J. Am. Chem. Soc., 1959, 81, 4600–4606 CrossRef CAS.
Footnotes |
| † Dedicated to our friend and mentor, Paul von Ragué Schleyer. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures, spectra, and X-ray data. CCDC 1452788 (1) and 1452902 (5). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc01309k |
| § We are currently performing a theoretical study on this topic which will be published separately. |
| ¶ M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision C.02, Gaussian, Inc., Wallingford CT, 2004. |
| This journal is © The Royal Society of Chemistry 2016 |