
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
CH3-deprotonation of 9-methylanthracene under mild conditions†‡
Nils
Finkelmeier
,
Arne
Visscher
,
Sebastian
Wandtke
,
Regine
Herbst-Irmer
and
Dietmar
Stalke
*
Institut für Anorganische Chemie der Universität Göttingen, Tammannstraße 4, 37077 Göttingen, Germany. E-mail: dstalke@chemie.uni-goettingen.de; Fax: +49-551-39-33459; Tel: +49-551-39-33000
First published on 18th March 2016
Abstract
The chromophore building-block 9-methylanthracene is selectively deprotonated at the methyl group and activated for reactions with electrophiles.
Introducing anthracene fluorophores into main group based sensors has been largely limited to the use of 9-lithioanthracene.1 The majority of luminescent compounds used in chemical sensing or organic light emission contain extended aromatic luminophore moieties connected to quencher moieties or molecular backbones via a spacer unit. Almost all synthetic approaches to sensing molecules described in the literature follow the established route to introduce alkyl spacers to the fluorophore via conventional organic synthesis, and then employing good leaving groups to introduce the receptor/quencher moiety. This process is almost entirely limited to SN-chemistry2 and brominated species.3 The umpolung option of the involved reactants using organometallics has not been established yet. In 2011 we reported the deprotonation of toluene under mild conditions using trimethylsilylmethyllithium.4 This gives the chance to deprotonate the sensor scaffold 9-methyl anthracene ((i) in Scheme 1) and convert the otherwise inert fluorophore moiety into a nucleophile. Reactions with quencher moieties QX would then transform the former methyl group of the starting material to a versatile methylene spacer in the reaction product ((ii) in Scheme 1).
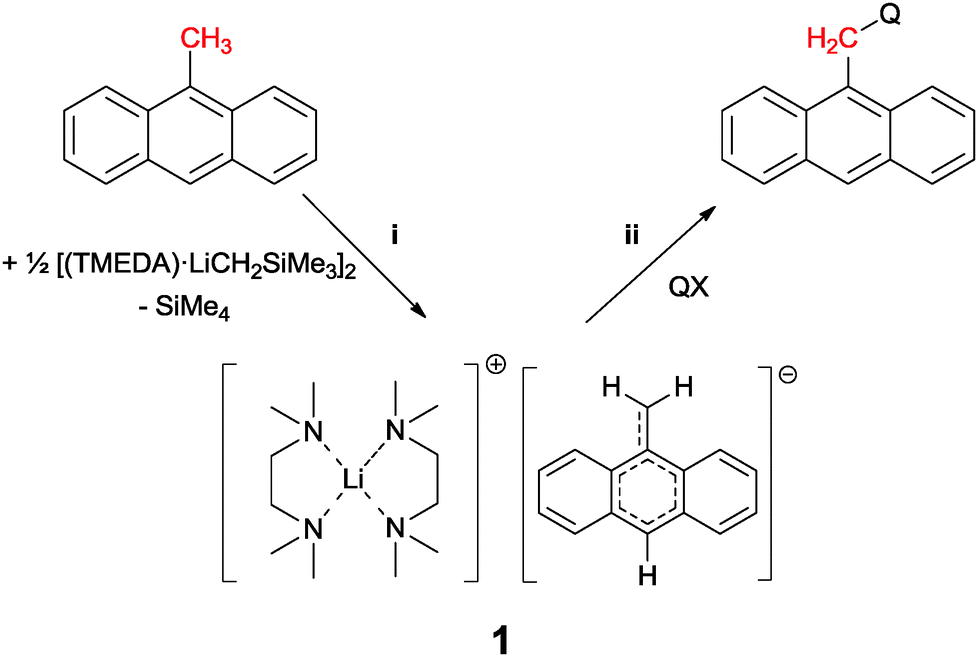 | ||
| Scheme 1 Deprotonation of 9-methylanthracene with [(TMEDA)LiCH2SiMe3]2 (i) and subsequent reaction with electrophile quencher moiety (ii). | ||
We embarked on that strategy by suspending 9-methyl anthracene in diethyl ether and added 1 eq. of TMEDA. The mixture was cooled to −15 °C and reacted with one eq. of [(TMEDA)LiCH2SiMe3]2 for 30 minutes. Upon addition of the organolithium component the color of the solution immediately changed from light yellow to dark green and was after completion close to black. Within minutes a dark crystalline precipitate was formed, leaving the mother liquor almost colorless. Due to the inevitably high speed of crystallization, the quality of the obtained crystals was poor and hampered the acquisition of good X-ray data.§
Nevertheless, the obtained results indicated a separated ion pair [Li(TMEDA)2] [H2C(C14H9)] (1) in the solid state (Fig. 1).
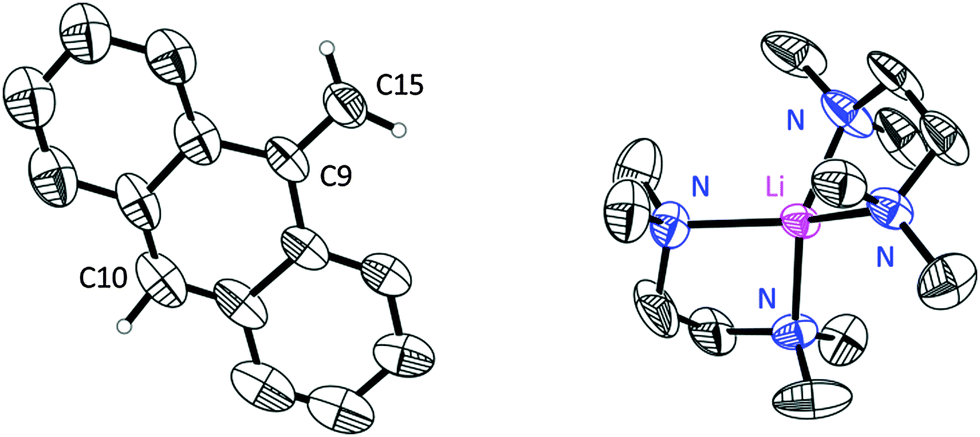 | ||
| Fig. 1 Crystal structure of one formula unit of 1a. The anisotropic displacement parameters are depicted at the 50% probability level. Disorder and hydrogen atoms besides those attached to C10 and C15 are omitted. | ||
The asymmetric unit contains one lithium ion and two half methyleneanthracene carbanions. The disorder could be resolved and the structure was modelled successfully. All hydrogen atoms could be found in the residual density map. The hydrogen atoms at position 10 and 15 were even refined freely (for more details see the ESI‡).
To avoid the instantaneous crystallization induced by the poor solubility of 1a in diethyl ether and to facilitate the hydrogen atom location, the reaction was cooled to and filtered at −78 °C. The lithiated compound was re-dissolved in pre-cooled THF. Only after several months at low temperature single crystals were obtained from the THF solution, which were again subjected to an X-ray diffraction experiment. The slower reaction and crystallization process proved to be beneficial for the crystal quality and a resolution of 0.74 Å was reached (1b).
As indicated by the cell parameters both structures 1a and 1b differ significantly. Although the lithium methyleneanthracenide also forms a separated ion pairs, the asymmetric unit contains an additional molecule of the MeC14H9 starting material, because [(TMEDA)LiCH2SiMe3]2 is always added in slightly deficient proportion to avoid dilithiation. Unfortunately the anthracene molecules as well as the TMEDA donor molecules are again disordered. An unambiguous assignment of the CH2/CH3 positions unfortunately was not possible (see ESI‡). Therefore we concentrate on the structure 1a. Although the crystal structures cannot ultimately answer all questions concerning the position of the negative charge, they do show that the deprotonation using [(TMEDA)LiCH2SiMe3]2 in fact works5 and that the obtained structure differs considerably from the structure of benzyl lithium,4b,c,6 which forms contact ion pairs to give either monomers,4c,6a,e cyclic tetramer,4c,6c or a branched octamer4b depending on the donor base added.
To further locate the position of the charge, 1 was subjected to NMR experiments. 1H, 13C, H–H COSY, 13C HSQC, 13C HMBC and 7Li experiments were conducted and the structure could fully be recovered. At first sight, it is striking that the entire 1H NMR spectrum of 1 appears to be shifted up-field (Fig. 2, left top & bottom) compared to 9-methylanthracene. The signals cover a substantially wider range in the spectrum of 1. The general up-field shift indicates stronger shielding of the protons. While the chemical shifts of 9-methylanthracene are in the expected range of aromatic protons, the shifts found for 1 are much lower than expected. The integrals of the 1H NMR spectrum confirm the observation derived from the crystal structure that the deprotonation exclusively occurred at the methyl group. The 7Li NMR spectrum shows a single peak at −2.3 ppm suggesting that there is only one single lithium species present in 1. This is in accordance with the results of the diffraction experiments. Li–C coupling could not be resolved, supporting the assumption that the separated ion pair from the solid state is maintained in solution. The 13C NMR chemical shifts of C15 also indicate the deprotonation of the methyl group; the shift from 13.0 ppm (9-methylanthracene) to 75.0 ppm (1) is substantial (Fig. 2, right). In comparison, the chemical shift of the ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2 carbon atom in benzyl lithium is only 31.7 ppm (tetramer) and 36.1 ppm (monomer).
H2 carbon atom in benzyl lithium is only 31.7 ppm (tetramer) and 36.1 ppm (monomer).
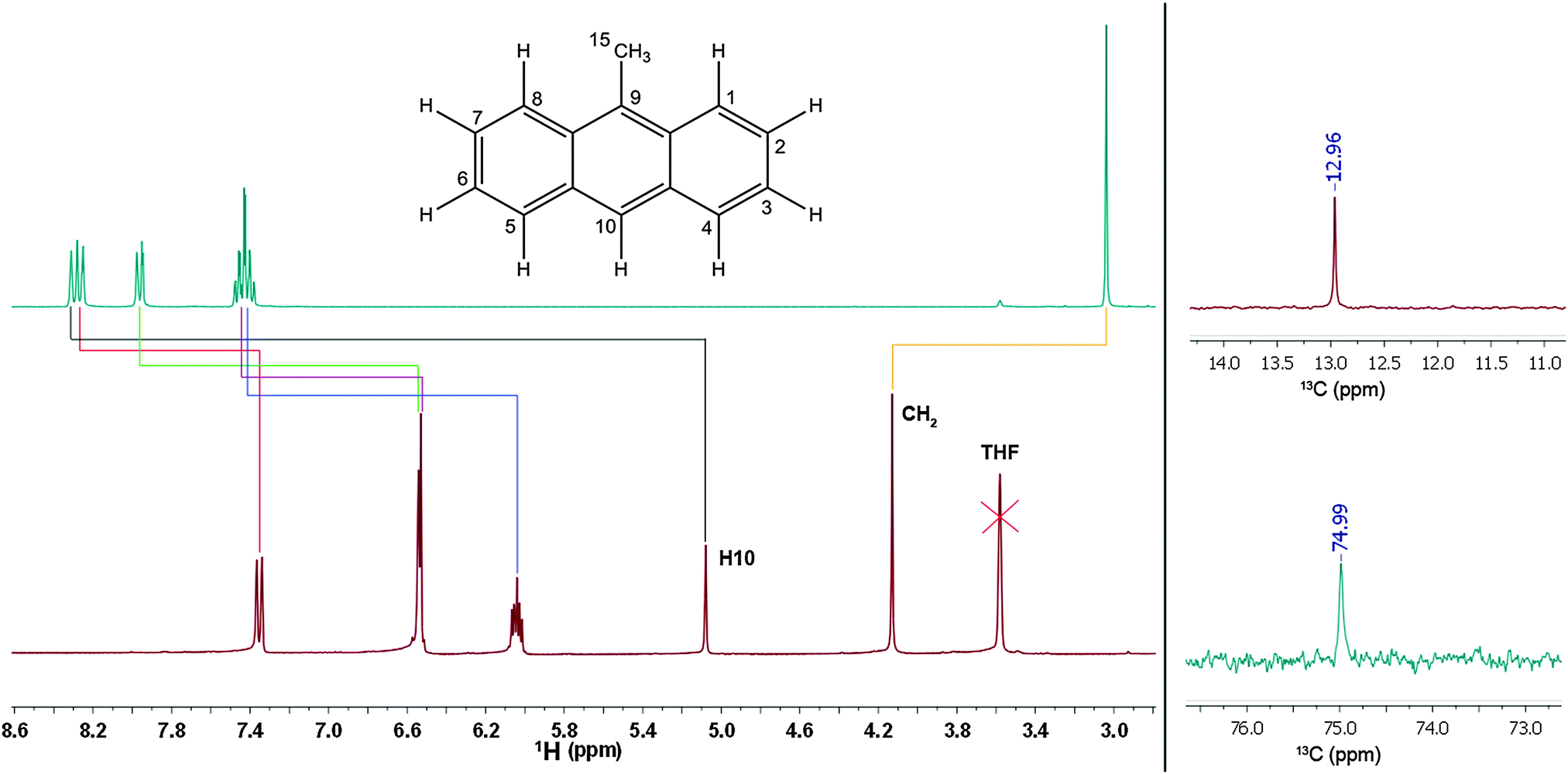 | ||
| Fig. 2 Left: 1H NMR signal assignment of methylanthracene (top) and 1 (bottom). Right: 13C NMR chemical shifts of C15 in methylanthracene (top) and 1 (bottom). | ||
The signal assignment between starting material and lithiated species depicted in Fig. 2 confirms that all signals are shifted up-field by deprotonation of the methyl group, except for the signal of the methylene protons themselves. Especially the shift of the H10-singlett from 8.33 ppm to 5.06 ppm is remarkable. The down-field shift of the methylene proton signal from 3.10 ppm to 4.12 ppm is quite surprising, because opposite observations were made for toluene/benzyl lithium, where a slight up-field shift of the methylene proton signal compared to the methyl proton signal of toluene was found.2c So far it deviates distinctly from the shift of 5.0 to 7.0 ppm expected for terminal vinylic protons. The up-field shift of the aromatic proton signals compared to toluene is, however, observed for benzyl lithium, although much weaker than in 1. In addition to the chemical shifts the 1JH,C coupling constants of C15 can be employed to determine the amount of delocalization at the benzyl anion. According to investigations of Boche et al.6a for a pyramidalized sp3 benzyl carbanion this coupling constant is expected to be 125 Hz, while the value expected for a planar sp2-CH2 group is 167 Hz. The 1JH,C coupling of 1 in solution of 154 Hz indicates a far higher sp2 character than in tetrameric benzyllithium [{Me2N(CH2)2OMe}(LiCH2C6H5)]4 (127 Hz) and monomeric [(PMDETA)(LiCH2C6H5)] (134 Hz). Taking into account the findings derived from the crystal structures (for details see ESI‡) and the NMR experiments, a localization of the charge coupling into the aromatic system can be assumed7 (b and c in Scheme 2), with only a minor contribution of the charge at C15 (a in Scheme 2). The down-field shift of the methylene protons is clearly too weak to postulate full π bonding between C9 and C15. Nevertheless, a fraction of the charge is transferred to the aromatic system, inducing an up-field shift of the corresponding proton signals.5 Single point DFT calculations (B3LYP/def2-TZVP) indicate that the planar sp2 methylene group substituted solvent separated ion pair is by 17 kJ mol−1 lower in energy than the sp3 pyramidal. The deprotonation of the inert 9-methylanthracene under mild conditions and the opportunity to further tune the reactivity by addition of various donor bases in the subsequent reactions with electrophiles clearly opens up new avenues in the syntheses of chromophores and sensor compounds, holding the inevitably important methylene bridge. Due to the accumulated charge almost exclusively at the deprotonated Cα, side products are not anticipated.
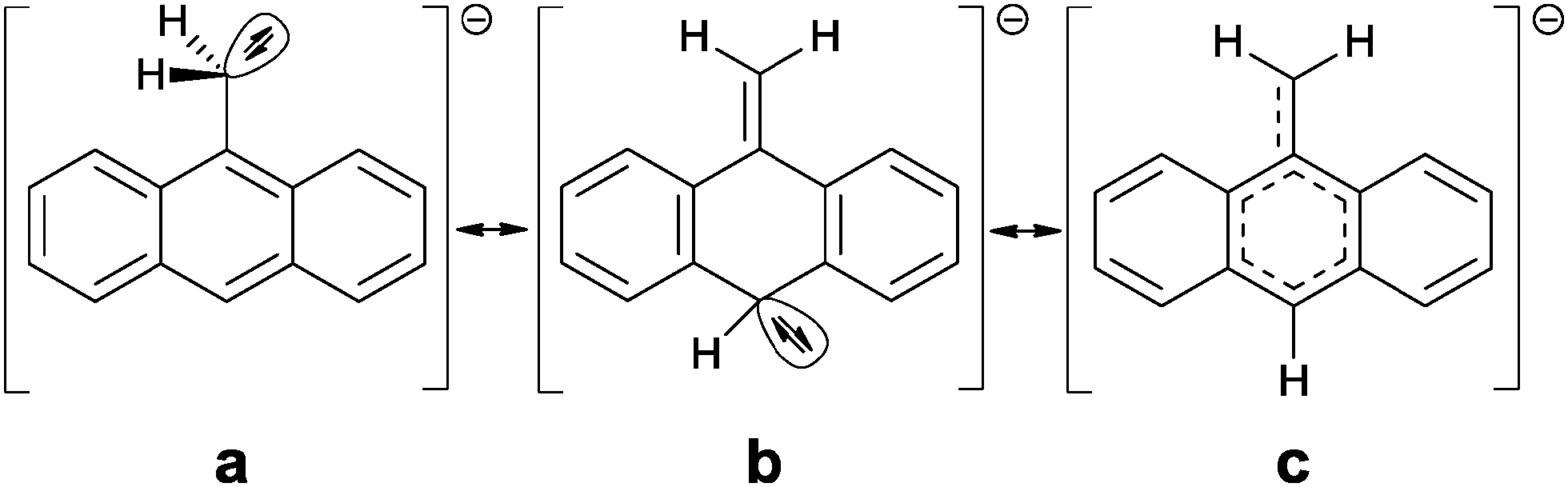 | ||
| Scheme 2 Possible delocalization of the negative charge. | ||
Notes and references
- C. A. Wade, A. E. J. Broomsgrove, S. Aldridge and F. Gabbaï, Chem. Rev., 2010, 110, 3958–3984 CrossRef CAS PubMed.
- (a) A. P. de Silva and S. Uchiyama, in Luminescence Applied in Sensor Science, ed. L. Prodi, M. Montalti and Z. Nelsi, Springer, Heidelberg, 2011 Search PubMed; (b) A. P. de Silva, T. P. Vance, M. E. S. West and G. D. Wright, Org. Biomol. Chem., 2008, 6, 2468–2481 RSC; (c) J. F. Callan, A. P. de Silva and D. C. Magri, Tetrahedron, 2005, 61, 8551–8588 CrossRef CAS.
- (a) D. Stern, N. Finkelmeier, K. Meindl, J. Henn and D. Stalke, Angew. Chem., 2010, 122, 7021–7024 ( Angew. Chem., Int. Ed. , 2010 , 49 , 6869–6872 ) CrossRef; (b) D. Stern, N. Finkelmeier and D. Stalke, Chem. Commun., 2011, 47, 2113–2115 RSC.
- (a) E. Carl and D. Stalke, Structure-Reactivity Relationship in Organolithium Compounds in Lithium Compounds in Organic Synthesis – From Fundamentals to Applications, ed. R. Luisi and V. Capriati, Wiley-VCH, Weinheim, 2014, pp. 1–31 Search PubMed; (b) T. Tatic, S. Hermann and D. Stalke, Organometallics, 2012, 31, 5615–5621 CrossRef CAS; (c) T. Tatic, S. Hermann, M. John, A. Loquet, A. Lange and D. Stalke, Angew. Chem., 2011, 123, 6796–6799 ( Angew. Chem., Int. Ed. , 2011 , 50 , 6666–6669 ) CrossRef.
- T. Tatic, H. Ott and D. Stalke, Eur. J. Inorg. Chem., 2008, 3765–3768 CrossRef CAS.
- e.g. (a) W. Zarges, M. Marsch, K. Harms and G. Boche, Chem. Ber., 1989, 122, 2303–2309 CrossRef CAS; (b) M. A. Beno, H. Hope, M. M. Olmstead and P. P. Power, Organometallics, 1985, 4, 2117–2121 CrossRef CAS; (c) D. R. Baker, W. Clegg, L. Horsburgh and R. E. Mulvey, Organometallics, 1994, 13, 4170–4172 CrossRef CAS; (d) D. Hoffmann, W. Bauer, F. Hampel, N. J. R. van Eikema Hommes, P. V. R. Schleyer, P. Otto, U. Pieper, D. Stalke, D. S. Wright and R. Snaith, J. Am. Chem. Soc., 1994, 116, 528–536 CrossRef CAS; (e) M. G. Davidson, D. Garcia-Vivo, A. R. Kennedy, R. E. Mulvey and S. D. Robertson, Chem. – Eur. J., 2011, 17, 3364–3369 CrossRef CAS PubMed.
- H. Ott, U. Pieper, D. Leusser, U. Flierler, J. Henn and D. Stalke, Angew. Chem., 2009, 121, 3022–3026 ( Angew. Chem., Int. Ed. , 2009 , 48 , 2978–2982 ) CrossRef.
- (a) T. Kottke and D. Stalke, J. Appl. Crystallogr., 1993, 26, 615–619 CrossRef; (b) D. Stalke, Chem. Soc. Rev., 1998, 27, 171–178 RSC.
- SAINT v7.68A in Bruker APEX v2011.9, Bruker AXS Inst. Inc., Madison, USA, 2008.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CAS.
- L. Krause, R. Herbst-Irmer and D. Stalke, J. Appl. Crystallogr., 2015, 48, 1907–1913 CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
Footnotes |
| † We are grateful to the DNRF funded Center of Materials Crystallography (DNRF93) and thank MSc Sebastian Heise for the calculations. |
| ‡ Electronic supplementary information (ESI) available: Crystallographic data, including the coordinates of all the species. CCDC 1452372 and 1452373. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc01261b |
| § Crystallographic details: shock cooled crystals were selected from a Schlenk under argon atmosphere using the X-TEMP2.8 The data were integrated with SAINT,9 and a multi-scan absorption correction (SADABS)10 was applied. A 3λ correction11 was applied. The structures were solved by direct methods (SHELXT)12 and refined on F2 using the full-matrix least-squares methods of SHELXL13 within the SHELXLE GUI.14 Crystal data for 1a: P21/c, a = 16.526(2), b = 9.874(2), c = 18.038(3) Å, β = 115.24(2)°, wR2 (all data) = 0.2106, R1 (I > 2σ(I)) = 0.0839, CCDC 1452372. Crystal data for 1b: P21/n, a = 9.486(2), b = 34.841(3), c = 19.391(2) Å, β = 93.77(2)°, wR2 (all data) = 0.1595, R1 (I > 2σ(I)) = 0.0545, CCDC 1452373. |
| This journal is © The Royal Society of Chemistry 2016 |