
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Long-chain NHC-stabilized RuNPs as versatile catalysts for one-pot oxidation/hydrogenation reactions†
L. M.
Martínez-Prieto‡
ab,
A.
Ferry‡
c,
L.
Rakers
c,
C.
Richter
c,
P.
Lecante
d,
K.
Philippot
*b,
B.
Chaudret
*a and
F.
Glorius
*c
aLPCNO; Laboratoire de Physique et Chimie des Nano-Objets, UMR5215 INSA-CNRS-UPS, Institut des Sciences appliquées, 135, Avenue de Rangueil, F-31077 Toulouse, France. E-mail: chaudret@insa-toulouse.fr
bLaboratoire de Chimie de Coordination, CNRS, LCC, 205, Route de Narbonne, F-31077 Toulouse, France. E-mail: karine.philippot@lcc-toulouse.fr
cOrganisch-Chemisches Institut, Westfälische Wilhelms-Universität Münster, Corrensstraße 40, 48149 Münster, Germany. E-mail: glorius@uni-muenster.de
dCEMES (Centre d’Elaboration de Matériaux et d’Etudes Structurales), CNRS, 29 Rue J. Marvig, F-31055 Toulouse, France
First published on 25th February 2016
Abstract
The synthesis and catalytic activity of long-chain NHC-stabilized RuNPs are presented. Full characterization of these novel nanostructures including surface state studies show that the ligand influences the number and the location of Ru active sites which impacts the NP catalytic activity, especially in hydrogenation reactions. The high stability and versatility of these nanosystems make them successful catalysts for both oxidation and hydrogenation reactions that can even be performed successively in a one pot-fashion.
The use of metal nanoparticles (MNPs) as catalysts has gained growing interest in the scientific community, with applications in a wide range of organic transformations such as hydrogenation, polymerization, oxidation and C–C coupling reactions.1 Their importance in catalysis comes from their large metallic surface area possessing numerous isolated active sites thus combining the benefits of homogeneous and heterogeneous catalysts. However, the high reactivity of unsupported NPs makes them, most of the time, very sensitive to air and moisture. The catalytic activity of MNPs is intensely affected by their size and the nature of the ligands used as stabilizers.2 Indeed, the stabilizing agents are capable of tuning the surface reactivity by modification of electronic and steric properties of the metallic surface.
N-Heterocyclic carbenes (NHCs) are among the most versatile ligands for transition metals in molecular coordination chemistry, broadly used in many catalytic reactions.3 Their use as ligands/stabilizers/modifiers of metal nanoparticles4–6 and flat metal surfaces7 has been explored recently. Some of us reported the use of NHC ligands to stabilize well-defined ruthenium nanoparticles (RuNPs) thanks to their electron-rich property and their capability to form strong metal ligand interactions.5c Moreover, some of us demonstrated that NHCs possessing long aliphatic chains in positions 4 and 5 of the imidazole ring increase the stability of palladium nanoparticles, forming a protective monolayer around the metallic surface.5h Based on these previous works, we opted to investigate the synthesis of two types of RuNPs stabilized with long aliphatic chain NHCs (LC-NHCs), containing either small or bulky substituents in positions 1 and 3, namely LC-IMe (1,3-dimethyl-4,5-diundecyl imidazoline) and LC-IPr (1,3-bis(2,6-diisopropylphenyl)-4,5-diundecyl imidazoline) (Fig. 1). With these two different LC-NHCs in hand, we evaluated the influence of the N-substituents on the size and reactivity of the RuNPs. The small methyl groups will minimize steric hindrance at the metallic surface, whereas diisopropylphenyl substituents are very bulky and might coordinate to the metal surface through van der Waals or π-interactions. We expected these novel RuNPs to possess high stability compared to those previously described. Their stability was investigated by TEM, HRTEM and WAXS analysis and their surface chemistry was probed by MAS-NMR and FT-IR after reaction with CO. The catalytic performance of these nanosystems was evaluated in hydrogenation and oxidation reactions including one-pot oxidation–hydrogenation processes. The route recently developed for RuNPs capped with non-isolable NHCs,5g was applied to prepare the long-chain NHC-stabilized RuNPs, namely Ru@LC-IPr and Ru@LC-IMe. Stable ruthenium colloidal solutions were hence obtained (Scheme 1) by the decomposition of the complex (1,5-cyclooctadiene)(1,3,5-cyclooctatriene)ruthenium [Ru(COD)(COT)] (THF; room temperature (r.t.); 3 bar H2) in the presence of 0.3 or 0.1 equivalent (equiv.) of LC-NHCs (LC-IMe or LC-IPr), previously generated in solution from their corresponding imidazolium salts. After purification by washing with MeOH, the RuNPs, obtained as a black powder, were re-dispersed in THF for Transmission Electronic Microscopy (TEM) analysis (Fig. 2). Ru@LC-NHCs prepared with 0.3 equiv., Ru@LC-IMe0.3 (1a) and Ru@LC-IPr0.3 (2a) reveal spherical, crystalline and well-dispersed NPs with a mean diameter of 1.3 (0.2) and 1.2 (0.3) nm, respectively and a narrow size dispersity. With 0.1 equiv. of LC-IPr Ru@LC-IPr0.1; (2b) larger NPs were obtained (1.5 (0.4) nm) with a higher size dispersity while Ru@LC-IMe0.1 (1b) presented a similar size to 1a (Fig. 2). High resolution TEM (HRTEM) micrographs of 1a (Fig. S1, ESI†) and 2a (Fig. S2, ESI†) indicate the presence of crystalline NPs displaying the hexagonal close packed (hcp) structure of bulk Ru. Wide-Angle X-ray Scattering (WAXS) analysis of 1a and 2a evidences the presence of unoxidized crystalline hcp RuNPs having a coherence length of ca. 1.3 nm, which is in accordance with HRTEM observations (Fig. S3 and S4, ESI†). 1a and 2a were characterized by infrared spectroscopy (FT-IR) and magic angle spinning solid-state NMR (MAS-NMR) with and without 1H–13C cross-polarization (CP) using the coordination of CO as a tool to locate the active sites on the Ru surface. Fig. S5 and S6 (see ESI†) show FT-IR spectra of 1a and 2a before (blue) and after (red) exposure to CO (1 bar, 20 h, r.t.). Remarkably, both FT-IR spectra exhibit the CO absorption band (between 1900 and 2000 cm−1) before the exposure to CO, which probably results from a decarbonylation process of THF during the NP synthesis, as recently observed.2d This evidences the high reactivity of 1a and 2a. In both cases, the coordination of CO was confirmed through the increase in intensity and shift to higher energy of the aforementioned band after CO exposure, specifically at 2011 cm−1 for 1a and 2012 cm−1 for 2a.
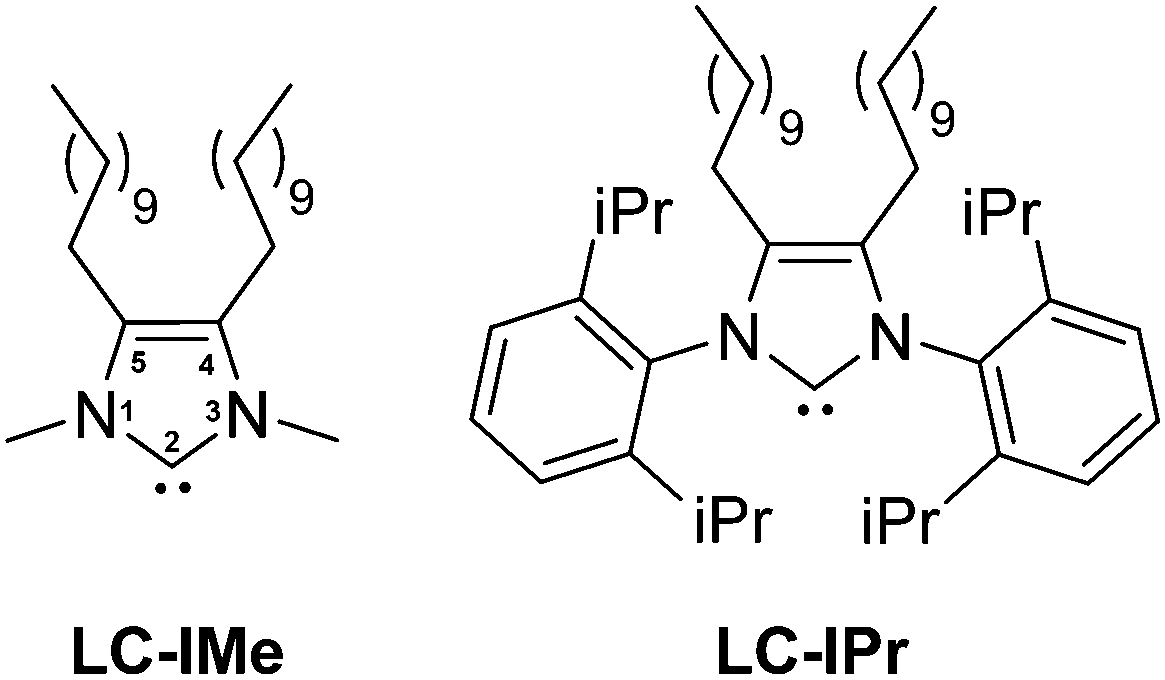 | ||
| Fig. 1 LC-IMe and LC-IPr long chain carbene ligands. | ||
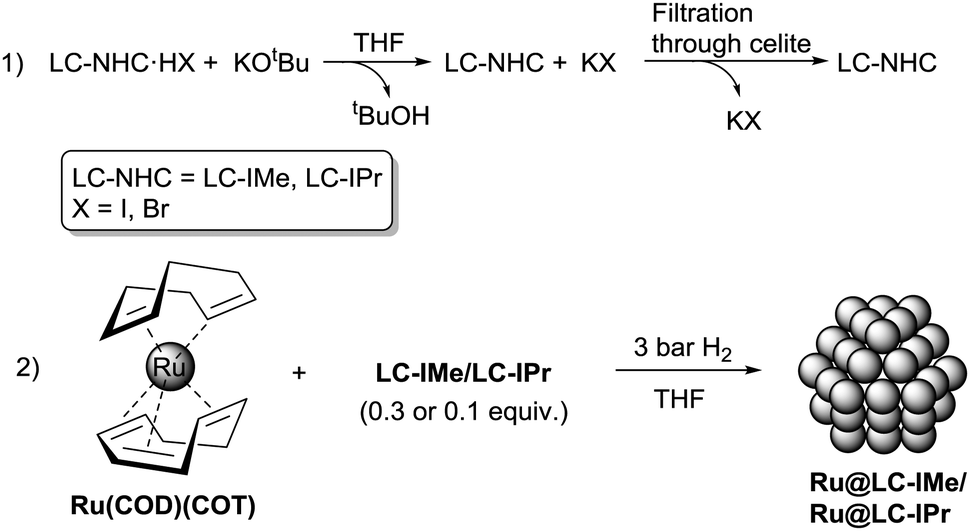 | ||
| Scheme 1 Synthesis of Ru@LC-IMe and Ru@LC-IPr. | ||
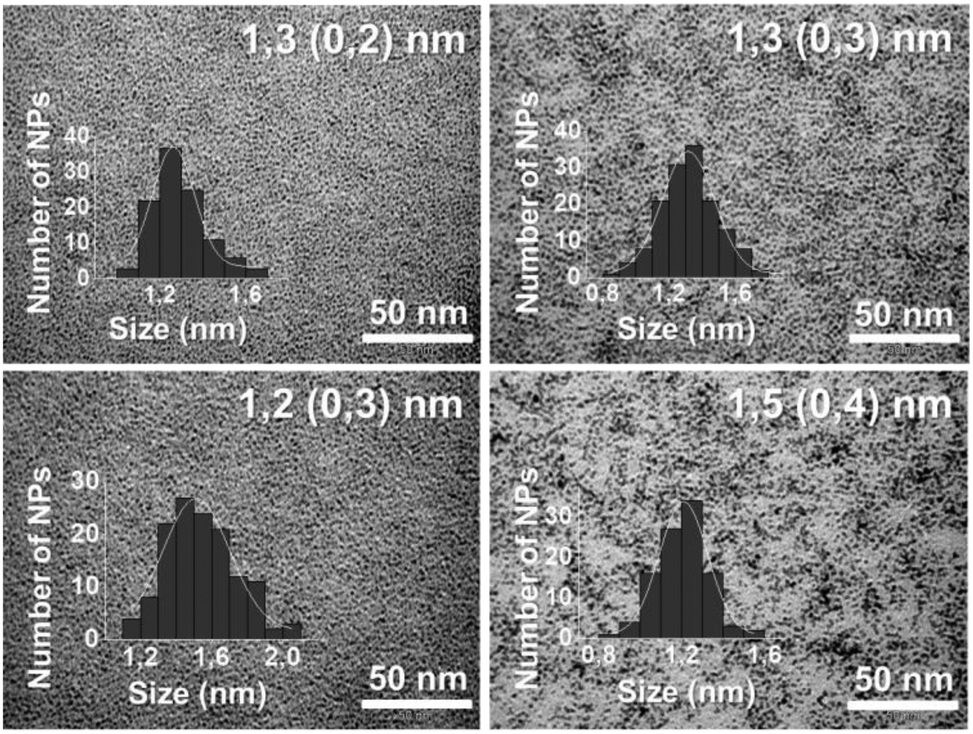 | ||
| Fig. 2 TEM micrographs and the corresponding size histograms of Ru@LC-IMe0.31a (left top), Ru@LC-IMe0.11b (right top), Ru@LC-IPr0.32a (left bottom) and Ru@LC-IPr0.12b (right bottom). | ||
The coordination of the LC-NHCs on the Ru surface was attested by 13C{1H} MAS-NMR. The spectra of 1a and 2a samples (bottom of Fig. S7 and S8, ESI†) present most of their characteristic peaks. The broad and low intensity signal near 120–130 ppm corresponds to the aromatic rings of the LC-IPr ligand and/or the imidazole backbone. The peaks at 29 ppm and 22 ppm belong to CH2 of the long alkyl chains as well as some alkyl groups from the nitrogen-bonded substituents (iPr or Me). The methyl groups at the extremity of the long alkyl chains are visible at 13 ppm for 1a and at 14 ppm for 2a. These signals are not visible on CP-MAS 13C{1H} NMR spectra (top of Fig. S7 and S8, ESI†) probably due to the high mobility of CH3 groups.
Coordination of CO was previously used to probe the NP metallic surface and it was proposed that COs coordinated in a bridging mode (COb) are located on the faces of the NPs and terminal COs (COt) on their apexes and edges.8 When 1a and 2a were reacted with 13CO (1 bar, r.t., 20 h), MAS 13C{1H} NMR spectra (Fig. 3) exhibited two new signals. The broad peak at δ ∼ 230 ppm can be assigned to COb and the sharper resonance at δ = 198–196 ppm to COt. In the 2a spectrum, the COb peak has a higher intensity than in the 1a one, evidencing as expected from the bulkiness of the ligand that 2a has more available face sites than 1a. In CP MAS, 13C{1H} spectra of 1a and 2a (Fig. S9, ESI†) the broad peak of COb is not visible and the intensity of COt peak decreases, potentially indicating that COb are not located close to LC-NHCs (hydrogen carriers) while COt are only partially located nearby the ligands. Interestingly, after reaction with CO, the peaks at 130–125 ppm (aromatic rings and imidazolium backbones) are not broad any more, probably because of their displacement from the surface by CO which allows them to tumble in the vicinity of the particle. All these data confirm that the RuNPs have different surface states depending on the ligand. In particular, 2a has more available face sites than 1a. We can thus expect different catalytic behavior (vide infra).
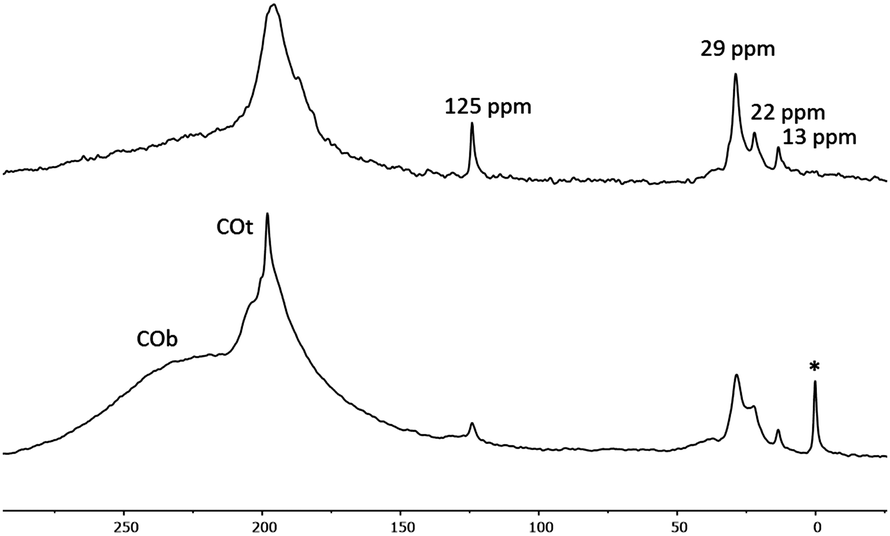 | ||
| Fig. 3 13C MAS NMR spectra of Ru@LC-IMe0.31a (top) and Ru@LC-IPr0.32a (bottom) after exposure to 13CO (1 bar, 20 h, at r.t.). The signal with asterisk corresponds to grease. | ||
NHC-stabilized RuNPs are efficient catalysts for the hydrogenation of arenes, ketones and olefins.5c,d,g Moreover, it was recently reported that LC-NHC-stabilized PdNPs present interesting chemoselectivities in the hydrogenation of olefins.5h Influence of this micelle-type NHC-backbone on the reactivity and chemoselectivity in hydrogenation (Table 1) as well as in oxidation reactions9 (Table 2) of the RuNPs was thus investigated. Significant differences of reactivity between the two nanosystems were observed in hydrogenation reactions. Indeed, neither ketone function nor the aromatic part were reduced with 1a under tested conditions (Table 1, entries 1, 3 and 5) whereas 2a mainly led to the over-reduced compound (Table 1, entries 2, 4 and 6). The change of LC-NHC loading from 0.3 to 0.1 equiv. makes the Ru@LC-IMe catalyst slightly more active (19% versus 0% in the hydrogenation of 3 leading mainly to 4). 1a and 1b having a similar size (1.3 nm), the difference of reactivity can be explained by the higher accessibility of the metallic surface of 1b, allowing a better diffusion of the substrate on the surface. In contrast to Ru@LC-IMe, the use of 2b led to a slight decrease of the reactivity in the hydrogenation of 3 (89% versus 100%), probably due to the larger size and the polydispersity of 2b compared to 2a. Following the same reactivity trend, acetamide 11 was converted into the corresponding amino-acid 12 only in the case of 2a (Table 1, entries 7 and 8). These experiments clearly show a high influence of the nitrogen-bonded substituents on the reactivity of the RuNPs in agreement with the number of free active sites on metallic surface (2a having more available face sites than 1a), whereas change of the loading of the ligand has only little influence.
| Entry/Ru-NP | Substrate | Cond. | Productsb | Conv.c (%) |
|---|---|---|---|---|
| a Reaction conditions: substrate (0.2 mmol), Ru-NPs (1 mg) under inert atmosphere. The pressure of H2, solvent, temperature and time are indicated in the table. b Determined by GC/MS. c Conversions were determined by GC/FID. | ||||
| 1/1a |
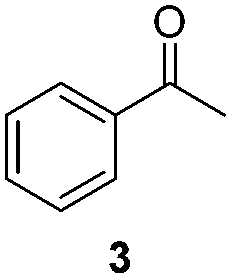
|
H2 (5 bar) hexane 16 h, 25 °C |
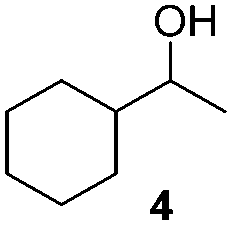
|
0 |
| 2/2a | 98 | |||
| 3/1a |
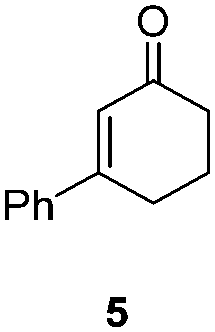
|
H2 (5 bar) toluene 16 h, 25 °C |
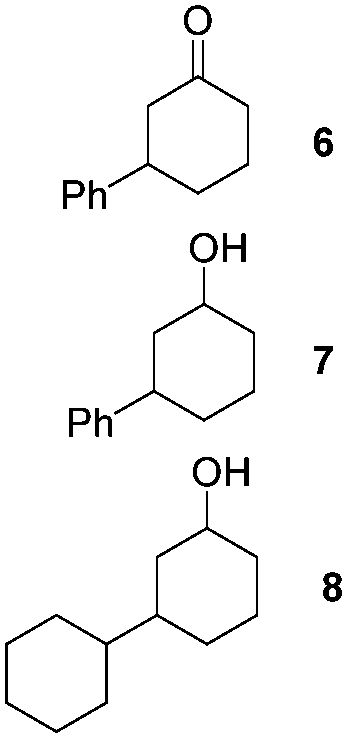
|
65 (only 6) |
| 4/2a | Quant (only 7 & 8:8 ≫ 7) | |||
| 5/1a |
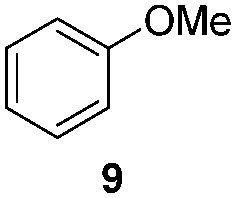
|
H2 (35 bar) hexane 16 h, 30 °C |
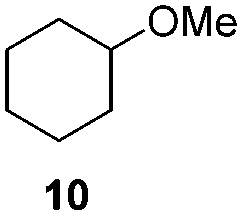
|
0 |
| 6/2a | Quant | |||
| 7/1a |
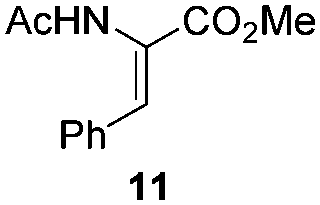
|
H2 (5 bar) toluene 16 h, 25 °C |
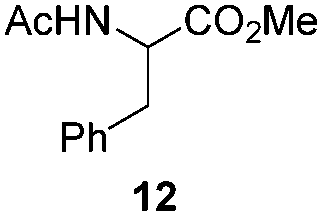
|
Trace |
| 8/2a | Quant | |||
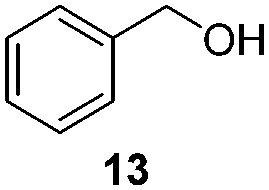
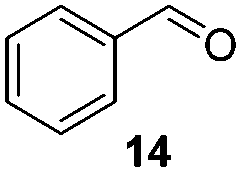
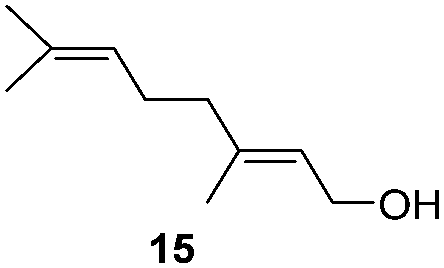
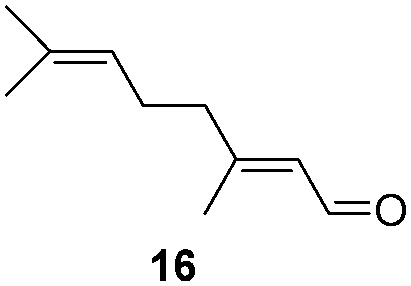
To test the stability of these NPs against oxygen, 1a and 2a were exposed to air for 3 days at r.t. In these conditions, only a partial oxidation was observed by WAXS (Fig. S3 and S4, ESI†). The oxide formed is mostly amorphous while the ruthenium core displays a small residual coherence length. Consequently, oxidation reactions using oxygen as oxidant were tested (Table 2). Oxidation of primary alcohols into aldehydes (Table 2, entries 1–4), and benzyl amine into benzonitrile (Table 2, entries 5 and 6) were performed with both catalysts. The order of activity again is 2a > 1a. 1b and 2b are less active (around 25% yield in the oxidation of 13 for both 1b and 2b and around 55% in the oxidation of 15). The lower reactivity of 1b and 2b can be explained by the lower quantity of LC-NHCs in 1b and 2b which makes them less stable under oxidative conditions. Successive oxidation reactions were performed complementary with 1a and 2a. After a first oxidation run (16 h) with 13, a second substrate was added (15) for another 24 h run under fresh O2. For both 1a and 2a a higher conversion for 13 was then observed as well as a good conversion for 15 (Scheme S1, stability experiments, ESI†), showing the stability of the system for at least 40 h. “Post-mortem” TEM and HRTEM analysis showed that the size and dispersion of both NPs did not change significantly after the first run despite a loss of their crystallinity (Fig. S10 and S11, ESI†), in accordance with previous WAXS analyses. Thus, the possibility to run successively oxidation and hydrogenation reactions in a one-pot process was investigated (Table 3). Oxidation conditions were first optimized to obtain quantitative conversions. Once the oxidation was complete (determined by GC/MS analysis), O2 atmosphere was replaced by H2 for further hydrogenation to take place. Thus aldehyde 19 was obtained in a one-pot process using 1a, via quantitative oxidation of 15 into 16 followed by hydrogenation predominantly leading to aldehyde 19 (Table 3, entry 1). Contrary to 1a, 2a mainly led to the over-reduced compound 21 (Table 3, entry 2). Likewise, amine 22 was obtained in one pot from 13. Indeed, intermediate 14, formed by oxidation of 13, reacted with isopropylamine to form the corresponding imine which was selectively reduced with 54% to quant conversion (Table 3, entries 3 and 4).
| Entry/Ru-NP | Subst. | Cond. | Prod.b | Conv.c,d (%) Select.d |
|---|---|---|---|---|
| a Reaction conditions: substrate (0.1 mmol) under inert atmosphere. The quantity of Ru-NPs, the pressure of H2 and O2, solvent, temperature and time are indicated in the table. b Determined by comparison of GC/MS data with commercial or synthesized references. c Conversions correspond to the hydrogenation step. d Conversions and selectivities were determined by GC-FID. | ||||
| 1/1a (1.0 mg) | 15 | (1) O2 (1 bar) PhCF3, 16 h, r.t. |
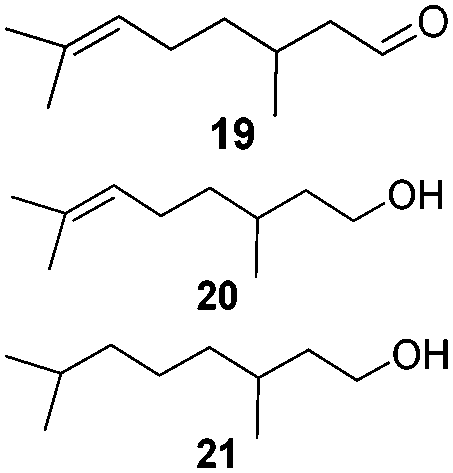
|
65
19/20/21:83/16/1 |
| 2/2a (1.0 mg) | (2) H2 (5 bar), r.t., 4 h |
Quant
19/20/21:0/8/92 |
||
| 3/1a (1.5 mg) | 13 | (1) O2 (1 bar) PhCF3, 16 h, r.t. |
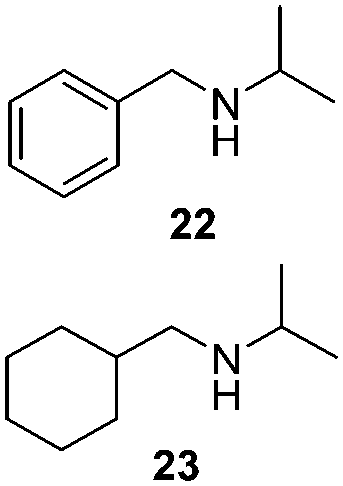
|
54
22/23:99/1 |
| 4/2a (1.5 mg) |
(2) Isopropyl-amine (1.5 equiv.), 2 h, r.t.
(3) H2 (5 bar), 45 °C, 16 h |
99
22/23:78/22 |
||
In summary, two long-chain NHC-stabilized RuNP samples were successfully synthesized and fully characterized. When tested in hydrogenation and oxidation reactions, these new Ru-NPs were shown active, thus attesting the presence of accessible active sites at their surface despite the long alkyl chains of the ligands. Interestingly, a clear influence of the ligand on selectivity and activity of the NPs was observed. Successful application of these RuNPs in one-pot oxidation–hydrogenation processes evidenced the value of these new systems of NPs as versatile air-stable catalysts for several organic transformations.
We thank CNRS, UPS-Toulouse, INSA, EU (ERC Advanced Grant, NANOSONWINGS 2009-246763) and the Deutsche Forschungsgemeinschaft (SFB 858 – Synergetic effects in chemistry) for financial support. We also thank V. Collière and L. Datas for TEM facilities (UMS-Castaing) and Y. Coppel (LCC) for NMR.
Notes and references
- (a) A. Roucoux, J. Schulz and H. Patin, Chem. Rev., 2002, 102, 3757 CrossRef CAS PubMed; (b) Nanoparticles and Catalysis, ed. D. Astruc, Wiley-VCH, Weinheim, 2008 Search PubMed; (c) Nanomaterials in Catalysis, ed. P. Serp and K. Philippot, Wiley-VCH, Weinheim, 2013 Search PubMed.
- (a) K. An and G. A. Somorjai, ChemCatChem, 2012, 4, 1512 CrossRef CAS; (b) D. González-Gálvez, P. Nolis, K. Philippot, B. Chaudret and P. W. N. M. van Leeuwen, ACS Catal., 2012, 2, 317 CrossRef; (c) L. M. Martínez-Prieto, S. Carenco, C. H. Wu, E. Bonnefille, S. Axnanda, Z. Liu, P. F. Fazzini, K. Philippot, M. Salmeron and B. Chaudret, ACS Catal., 2014, 4, 3160 CrossRef; (d) L. M. Martínez-Prieto, C. Urbaneja, P. Palma, J. Campora, K. Philippot and B. Chaudret, Chem. Commun., 2015, 51, 4647 RSC.
- (a) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485 CrossRef CAS PubMed; (b) N-Heterocyclic Carbenes in Synthesis, ed. S. P. Nolan, Wiley-VCH, New York, 2006 Search PubMed; (c) N-Heterocyclic Carbenes in Transition Metal Catalysis, ed. F. Glorius, Springer, Berlin, 2007 Search PubMed; (d) E. A. B. Kantchev, C. J. O’Brien and M. G. Organ, Angew. Chem., Int. Ed., 2007, 46, 2768 CrossRef CAS PubMed; (e) S. Würtz and F. Glorius, Acc. Chem. Res., 2008, 41, 1523 CrossRef PubMed; (f) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122 CrossRef CAS PubMed; (g) S. Diez-Gonzalez, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612 CrossRef CAS PubMed; (h) T. Dröge and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 6940 CrossRef PubMed; (i) N-Heterocyclic Carbenes in Transition Metal Catalysis, ed. C. S. J. Cazin, Springer, Berlin, 2011 Search PubMed.
- Review on the use of NHCs for nanoparticles and surfaces: A. V. Zhukhovitskiy, M. J. MacLeod and J. A. Johnson, Chem. Rev., 2015, 115, 11503 CrossRef CAS PubMed.
- For NHCs as ligands for unsupported NPs see for example: (a) J. Vignolle and T. D. Tilley, Chem. Commun., 2009, 7230 RSC; (b) E. C. Hurst, K. Wilson, I. J. S. Fairlamb and V. Chechik, New J. Chem., 2009, 33, 1837 RSC; (c) P. Lara, O. Rivada-Wheelaghan, S. Conejero, R. Poteau, K. Philippot and B. Chaudret, Angew. Chem., Int. Ed., 2011, 50, 12080 CrossRef CAS PubMed; (d) D. Gonzalez-Galvez, P. Lara, O. Rivada-Wheelaghan, S. Conejero, B. Chaudret, K. Philippot and P. W. N. M. van Leeuwen, Catal. Sci. Technol., 2013, 3, 99 RSC; (e) E. A. Baquero, S. Tricard, J. C. Flores, E. de Jesús and B. Chaudret, Angew. Chem., Int. Ed., 2014, 53, 13220 CrossRef CAS PubMed; (f) A. Ferry, K. Schaepe, P. Tegeder, C. Richter, K. M. Chepiga, B. J. Ravoo and F. Glorius, ACS Catal., 2015, 5, 5414 CrossRef CAS; (g) L. M. Martínez-Prieto, A. Ferry, P. Lara, C. Richter, K. Philippot, F. Glorius and B. Chaudret, Chem. – Eur. J., 2015, 21, 17495 CrossRef PubMed; (h) C. Richter, K. Schaepe, F. Glorius and B. J. Ravoo, Chem. Commun., 2014, 50, 3204 RSC ; For the application of these long chain NHCs in micellar catalysis, see:; (i) A. Rühling, H.-J. Galla and F. Glorius, Chem. – Eur. J., 2015, 21, 12291 CrossRef PubMed.
- For NHCs on supported metal NPs, see: (a) K. V. S. Ranganath, J. Kloesges, A. H. Schäfer and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 7786 CrossRef CAS PubMed; (b) K. V. S. Ranganath, A. Schäfer and F. Glorius, ChemCatChem, 2011, 3, 1889 CrossRef CAS; (c) D. Yu, M. X. Tan and Y. Zhang, Adv. Synth. Catal., 2012, 354, 969 CrossRef CAS.
- For NHCs on metal surfaces, see: (a) T. Weidner, N. Ballav, U. Siemeling, D. Troegel, T. Walter, R. Tacke, D. G. Castner and M. Zharnikov, J. Phys. Chem. C, 2009, 113, 19609 CrossRef CAS PubMed; (b) T. Weidner, J. E. Baio, A. Mundstock, C. Große, S. Karthäuser, C. Bruhn and U. Siemeling, Aust. J. Chem., 2011, 64, 1177 CrossRef CAS PubMed; (c) A. V. Zhukhovitskiy, M. G. Mavros, T. V. Voorhis and J. A. Johnson, J. Am. Chem. Soc., 2013, 135, 7418 CrossRef CAS PubMed; (d) C. M. Crudden, J. H. Horton, I. I. Ebralidze, O. V. Zenkina, A. B. McLean, B. Drevniok, Z. She, H.-B. Kraatz, N. J. Mosey, T. Seki, E. C. Keske, J. D. Leake, A. Rousina-Webb and G. Wu, Nat. Chem., 2014, 6, 409 CrossRef CAS PubMed.
- F. Novio, K. Philippot and B. Chaudret, Catal. Lett., 2010, 140, 1 CrossRef CAS.
- (a) M. Tristany, K. Philippot, Y. Guari, V. Collière, P. Lecante and B. Chaudret, J. Mater. Chem., 2010, 20, 9523 RSC; (b) E. J. García-Suárez, M. Tristany, A. B. García, V. Collière and K. Philippot, Microporous Mesoporous Mater., 2012, 153, 155 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc01130f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |