
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Distinguishing N-acetylneuraminic acid linkage isomers on glycopeptides by ion mobility-mass spectrometry†
H.
Hinneburg‡
ab,
J.
Hofmann‡
bc,
W. B.
Struwe
d,
A.
Thader
e,
F.
Altmann
 e,
D.
Varón Silva
a,
P. H.
Seeberger
ab,
K.
Pagel
*bc and
D.
Kolarich
*a
e,
D.
Varón Silva
a,
P. H.
Seeberger
ab,
K.
Pagel
*bc and
D.
Kolarich
*a
aDepartment of Biomolecular Systems, Max Planck Institute of Colloids and Interfaces, 14424 Potsdam, Germany. E-mail: daniel.kolarich@mpikg.mpg.de
bDepartment of Biology, Chemistry, Pharmacy, Freie Universität Berlin, 14195 Berlin, Germany. E-mail: kevin.pagel@fu-berlin.de
cFritz Haber Institute of the Max Planck Society, 14195 Berlin, Germany
dDepartment of Chemistry, Physical and Theoretical Chemistry Laboratory, University of Oxford, OX1 3QZ, Oxford, UK
eDepartment of Chemistry, University of Natural Resources and Applied Life Sciences, Vienna, Austria
First published on 24th February 2016
Abstract
Differentiating the structure of isobaric glycopeptides represents a major challenge for mass spectrometry-based characterisation techniques. Here we show that the regiochemistry of the most common N-acetylneuraminic acid linkages of N-glycans can be identified in a site-specific manner from individual glycopeptides using ion mobility-mass spectrometry analysis of diagnostic fragment ions.
Protein glycosylation as post-translational modification tremendously influences cellular events such as cell–cell interactions and receptor recognition.1,2 Glycosylation is highly dynamic, cell-type specific and depends on a variety of additional factors such as the developmental status of the cell.3,4 Understanding the impact of glycosylation on protein function requires detailed knowledge of individual glycan structures and their site-specific distribution, as glycoprotein macro- and microheterogeneity can vary tremendously within a single protein.5–9 Therefore, knowledge of both individual glycan structure and the location on a given protein is important in revealing these complex structure–function relationships.6
Liquid chromatography-mass spectrometry (LC-MS) is a very sensitive technique that is widely used for studying site-specific protein glycosylation.5,7,9,10 Differentiation of minor changes in glycan structure, such as terminal N-acetylneuraminic acid (NeuAc) linkages, directly from glycopeptides is very challenging due to limits in LC separation and the isobaric nature of the fragments observed by MS.7,10 A promising technique capable of providing additional structural information is ion mobility spectrometry (IM) coupled to mass spectrometry (IM-MS).11 In IM, analyte ions travel through a cell filled with an inert neutral gas aided by a weak electric field and undergo a series of low-energy collisions with the gas. Compact ions undergo fewer collisions with the drift gas than more extended ions and therefore traverse the IM cell faster. As a result, in IM-MS molecular ions or their fragments are not only separated according to their mass and charge, but also according to their size and shape, which enables the separation of isomers. The obtained drift times can be converted into rotationally-averaged collision cross sections (CCSs),12,13 which are absolute biophysical properties that can be used for structural classification by reference to database values.14
IM-MS studies on isolated glycan isomers show great promise,15–19 however, very few reports focus on isomeric glycopeptides.18,20 Here we report a universally applicable and rapid approach capable of differentiating α2,3 and α2,6 NeuAc linkages in N-glycopeptides without any additional sample preparation steps. Using IM-MS we evaluated a small set of well-defined, synthetic glycopeptides carrying N-glycans containing either α2,3 or α2,6 linked NeuAc residues. To illustrate the robustness of the method, we tested complex mixtures using two forms of α-1 proteinase inhibitor (A1PI) produced in different cell types.
Homogeneous glycopeptides were generated by chemical and chemo-enzymatic synthesis for systematic IM-MS method development. An asparagine (Asn) building block carrying biantennary, mono- or disialylated N-glycans was obtained from egg yolk using a combination of extraction and proteolytic digestion steps (for details see ESI†).21,22 This glycan-Asn building block was Fmoc protected, the NeuAc residues benzylated and the molecule subsequently used to synthesize N-glycopeptides by solid-phase peptide synthesis (SPPS).
The first set of glycopeptides was designed based on the naturally occurring tryptic peptide sequence from human butyrylcholinesterase (505YGNPNETQNNSTSWPVFK522, UniProt entry P06276).23 This peptide contains three possible glycosylation sites (boldface) defined by the consensus sequence N-X-T/S/C (X ≠ P), two of which are glycosylated in serum.23 To reduce complexity, the sequence was simplified to YGNVNETQNNSFK and an α2,6 disialylated, biantennary glycan was selectively incorporated by SPPS at one glycosylation site, either near the N-(GP1) or near the C-terminus (GP2). IM-MS experiments were performed to determine whether the method can separate the isobaric glycopeptides. As protonated ions, both isomers, regardless of the charge state, could not be separated and showed identical drift times (Fig. S4, ESI†). However, when quadruply deprotonated ions ([M − 4H]4− = 928) were measured, GP1 and GP2 had noticeably different drift times of 5.80 and 5.33 ms (Fig. 1). Both isomers, when examined as mixtures, were nearly baseline separated illustrating that IM-MS can in principle be used to differentiate isobaric glycopeptides that merely differ in their glycosylation site.
 | ||
| Fig. 1 IM-MS separation of the isobaric glycopeptides GP1 and GP2. Two isomeric glycopeptides that share the same sequence and attached glycan, but differ in the site of glycan attachment can be distinguished based on their drift time (top and middle) and separated in mixtures (bottom) when analysed as quadruply deprotonated ions.§ | ||
Next, we examined two glycopeptides with a single glycosylation site but different glycan structures. Specifically, the attached complex-type glycans differed in the linkage of the terminal NeuAc residue, that is either α2,3- or α2,6-linked to galactose. Subtle differences in NeuAc regiochemistry are of biological and biopharmaceutical importance8,24 and are challenging to characterise using established glycoproteomics techniques.25,26 The investigated peptides are designed based on the human protein C fragment 284EVFVHPNYSK293 (UniProt entry P04070) that contains one glycosylation site (boldface). The peptide was synthesized by SPPS using an Asn building block containing an α2,6 monosialylated, biantennary N-glycan. Subsequently, a fraction of the resulting glycopeptide (GP3) was desialylated using trifluoroacetic acid followed by enzymatic re-sialylation using recombinant β-galactoside α2,3-sialyltransferase 3 (see ESI†). Finally, a glycopeptide (GP4) exclusively carrying α2,3 linked NeuAc residues was obtained.
Only marginal, non-significant drift time differences between GP3 and GP4 were observed for 3+ molecular ions of the intact glycopeptides such that they cannot be separated in mixtures (Fig. 2b). Given the minor structural differences in the glycan moiety compared to the overall size of the molecule this result was not surprising.
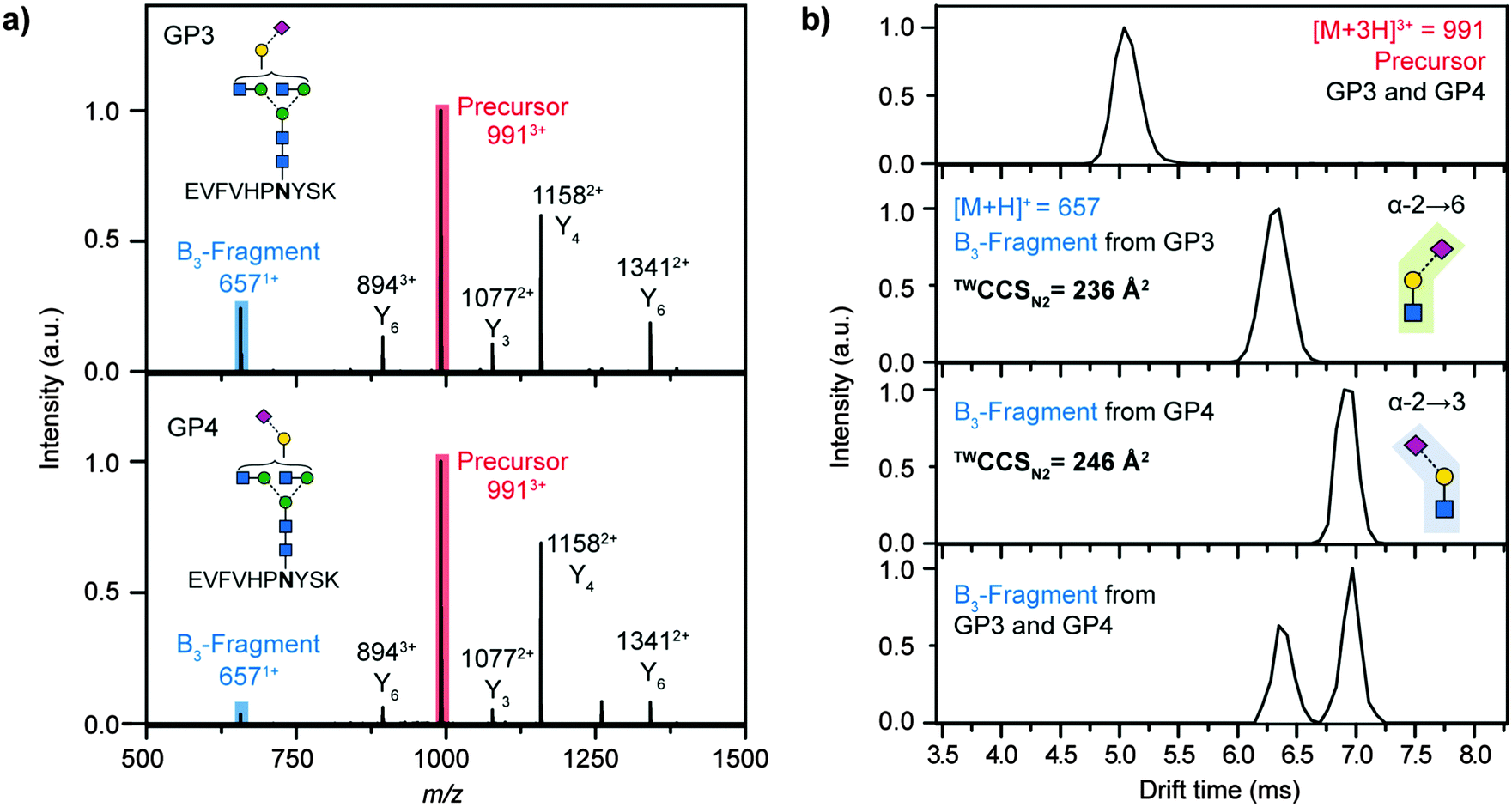 | ||
| Fig. 2 Differentiation of N-acetylneuraminic acid (NeuAc) linkage isomers using CID fragmentation and subsequent IM-MS analysis. Two isomeric glycopeptides, which either carry α2,6 (GP3) or α2,3 (GP4) linked NeuAc were analysed. (a) Both peptides exhibit identical MS/MS spectra, as shown for the triply protonated precursor ion (red).¶ (b) When analysed as mixture the intact glycopeptide ions cannot be separated by IM-MS (m/z 991, red). B3-trisaccharide fragments (m/z 657, blue) directly cleaved from the glycopeptide by CID on the other hand show characteristic drift times depending on the regiochemistry of the NeuAc linkage, which allows unambiguous identification α2,3 and α2,6 linked isoforms.§ | ||
When collision-induced dissociation (CID) is applied to positively charged glycopeptide ions, glycosidic cleavages are the preferred fragmentation pathway resulting in a multitude of oligosaccharide-only fragment ions (B- and Y-type fragments in Fig. 2).¶![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 27 Tandem MS analysis of GP3 and GP4 yielded almost identical CID fragment spectra and did not provide diagnostic information for any glycan structural features (Fig. 2a). However, from these oligosaccharide-only fragment ions the m/z 657 B3 type fragment is of particular interest because this oxonium ion corresponds to a trisaccharide consisting of Gal, GlcNAc and NeuAc residues in N-glycopeptides. To elucidate whether regiochemical differences of the NeuAc linkage in GP3 and GP4 leads to drift time differences of the resulting fragments, CID experiments were followed by IM-MS analysis. The extracted arrival time distributions (ATDs) of these m/z 657 B3 ions were vastly different for GP3 and GP4 (Fig. 2b), with the α2,6 fragment exhibiting a considerably shorter drift time when compared to the α2,3 equivalent. When both glycopeptides were mixed, the isomeric NeuAc-containing fragments showed baseline separation. In addition, the collision cross sections in nitrogen drift gas (TWCCSN2) of 236 Å2 for the α2,6 linked NeuAc and 246 Å2 for the α2,3 linked NeuAc fragments differed about 4%, well above the 1.5% error of the method.13 These values are highly diagnostic to the regiochemistry of the underlying NeuAc linkage and can be used to gain site-specific information on important glycan structural features directly from individual glycopeptides in a single experiment.
27 Tandem MS analysis of GP3 and GP4 yielded almost identical CID fragment spectra and did not provide diagnostic information for any glycan structural features (Fig. 2a). However, from these oligosaccharide-only fragment ions the m/z 657 B3 type fragment is of particular interest because this oxonium ion corresponds to a trisaccharide consisting of Gal, GlcNAc and NeuAc residues in N-glycopeptides. To elucidate whether regiochemical differences of the NeuAc linkage in GP3 and GP4 leads to drift time differences of the resulting fragments, CID experiments were followed by IM-MS analysis. The extracted arrival time distributions (ATDs) of these m/z 657 B3 ions were vastly different for GP3 and GP4 (Fig. 2b), with the α2,6 fragment exhibiting a considerably shorter drift time when compared to the α2,3 equivalent. When both glycopeptides were mixed, the isomeric NeuAc-containing fragments showed baseline separation. In addition, the collision cross sections in nitrogen drift gas (TWCCSN2) of 236 Å2 for the α2,6 linked NeuAc and 246 Å2 for the α2,3 linked NeuAc fragments differed about 4%, well above the 1.5% error of the method.13 These values are highly diagnostic to the regiochemistry of the underlying NeuAc linkage and can be used to gain site-specific information on important glycan structural features directly from individual glycopeptides in a single experiment.
To evaluate whether similar diagnostic fragments could be obtained from complex mixtures within a glycoproteomics workflow, forms of the glycoprotein A1PI were tested (Fig. 3a). N-Glycan NeuAc linkages on A1PI differ depending on the biological source of the protein. Human plasma A1PI contains mostly α2,6 linked NeuAc residues25 whereas the same protein recombinantly expressed in Chinese hamster ovary (CHO) cells only contains α2,3 sialylated glycans.28 A1PI samples from both sources were purified using SDS-gel electrophoresis followed by in-gel tryptic digestion and glycopeptide enrichment using hydrophilic interaction chromatography (HILIC) to remove unglycosylated peptides as these can suppress efficient glycopeptide detection (for details see ESI†).22 Subsequently, the purified glycopeptides were analysed off-line by IM-MS (Fig. S5 and S6, ESI†). Known glycopeptide precursor ions25 were m/z-selected for tandem IM-MS experiments and characteristic B3 fragments were observed after CID (Fig. 3b). Importantly, the drift times of the obtained B3 fragments of 7.03 ms (from CHO precursor m/z 1273 and 1401) and 6.44 ms (from human plasma precursor m/z 1321) were essentially identical to those observed for the synthetic glycopeptides GP4 (6.96 ms) and GP3 (6.38 ms), respectively. In addition, the TWCCSN2 of the oxonium fragment ions (Fig. 3b) were consistent with the synthetic reference glycopeptide data and can be used to differentiate α2,3 from α2,6 sialylated N-glycans from all sialylated glycopeptides obtained from CHO-derived and human A1PI (Fig. 3b). The universal applicability of this approach is demonstrated, as size and sequence of the glycopeptide precursors did not affect the drift time of the resulting B3 fragment. This finding underscores the diagnostic nature of the α2,3 or α2,6 fragments and justifies this approach as a reliable fragment-based method to obtain N-glycan structure information directly from glycopeptides.
 | ||
| Fig. 3 Regiochemistry analysis of N-acetylneuraminic acid (NeuAc) linkages in α1-proteinase inhibitor (A1PI). (a) A1PI isolated from human plasma and recombinantly expressed in Chinese hamster ovary (CHO) cells was purified, digested with trypsin, and the glycopeptides HILIC-enriched. (b) Fragmentation of the obtained glycopeptides and subsequent IM-MS analysis of the characteristic B3-trisaccharide fragments (m/z 657) enabled the differentiation of α2,3 from α2,6 linked NeuAc. The observed fragment drift times and TWCCSN2 are independent of the underlying precursor sequence.§ | ||
In conclusion, we show that IM-MS can significantly improve the identification of isomeric glycopeptides and fits seamlessly within existing glycoproteomics workflows. Peptides with two distinct glycosylation sites can be differentiated directly as intact molecular ions using IM-MS. The regiochemistry of the prevalent α2,3 and α2,6 NeuAc linkages in N-glycosylated peptides can be distinguished and allows for the unambiguous identification in a site-specific manner on basis of the CCSs of diagnostic B3-type fragments that are cleaved directly from mass-selected glycopeptide precursors. The approach is fast, does not require derivatisation and is universally applicable regardless of the nature of the investigated glycoprotein. Our data highlight the immense potential of IM-MS to be implemented into existing glycoproteomic workflows.
We thank the Max Planck Society for generous financial support, ProBioGen AG for kindly providing A1PI produced in CHO cells and Daniel Maresch for technical assistance in enzymatic glycan remodelling. DK and HH acknowledge support by the European Union (Seventh Framework Programme “Glycoproteomics” project, grant number PCIG09-GA-2011-293847 and IBD-BIOM project grant number 305479). DVS acknowledges the RIKEN-Max Planck Joint Center for Systems Chemical Biology for financial support. WBS thanks the Biotechnology and Biological Sciences Research Council (BBSRC) [BB/L017733/1] for financial support. JH and KP thank Dr. Gert von Helden and Prof. Michael T. Bowers for fruitful discussions and support.
Notes and references
- P. R. Crocker, J. C. Paulson and A. Varki, Nat. Rev. Immunol., 2007, 7, 255–266 CrossRef CAS PubMed.
- A. Grigorian, S. Torossian and M. Demetriou, Immunol. Rev., 2009, 230, 232–246 CrossRef CAS PubMed.
- Essentials of Glycobiology, ed. A. Varki, R. D. Cummings, J. D. Esko, H. H. Freeze, P. Stanley, C. R. Bertozzi, G. W. Hart and M. E. Etzler, Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY), USA, 2nd edn, 2009 Search PubMed.
- K. W. Moremen, M. Tiemeyer and A. V. Nairn, Nat. Rev. Mol. Cell Biol., 2012, 13, 448–462 CrossRef CAS PubMed.
- R. Plomp, P. J. Hensbergen, Y. Rombouts, G. Zauner, I. Dragan, C. A. M. Koeleman, A. M. Deelder and M. Wuhrer, J. Proteome Res., 2014, 13, 536–546 CrossRef CAS PubMed.
- K.-T. C. Shade, B. Platzer, N. Washburn, V. Mani, Y. C. Bartsch, M. Conroy, J. D. Pagan, C. Bosques, T. R. Mempel, E. Fiebiger and R. M. Anthony, J. Exp. Med., 2015, 212, 457–467 CrossRef CAS PubMed.
- H. Hinneburg, K. Stavenhagen, U. Schweiger-Hufnagel, S. Pengelley, W. Jabs, P. H. Seeberger, D. V. Silva, M. Wuhrer and D. Kolarich, J. Am. Soc. Mass Spectrom., 2016, 27, 507–519 CrossRef CAS PubMed.
- J. Stadlmann, M. Pabst and F. Altmann, J. Clin. Immunol., 2010, 30, 15–19 CrossRef PubMed.
- I. Loke, N. H. Packer and M. Thaysen-Andersen, Biomolecules, 2015, 5, 1832–1854 CrossRef CAS PubMed.
- K. Stavenhagen, R. Plomp and M. Wuhrer, Anal. Chem., 2015, 87, 11691–11699 CrossRef CAS PubMed.
- B. C. Bohrer, S. I. Merenbloom, S. L. Koeniger, A. E. Hilderbrand and D. E. Clemmer, Annu. Rev. Anal. Chem., 2008, 1, 293–327 CrossRef CAS PubMed.
- K. Pagel and D. J. Harvey, Anal. Chem., 2013, 85, 5138–5145 CrossRef CAS PubMed.
- J. Hofmann, W. B. Struwe, C. A. Scarff, J. H. Scrivens, D. J. Harvey and K. Pagel, Anal. Chem., 2014, 86, 10789–10795 CrossRef CAS PubMed.
- W. B. Struwe, K. Pagel, J. L. P. Benesch, D. J. Harvey and M. P. Campbell, Glycoconjugate J., 2015 DOI:10.1007/s10719-015-9613-7.
- W. Gabryelski and K. L. Froese, J. Am. Soc. Mass Spectrom., 2003, 14, 265–277 CrossRef CAS PubMed.
- B. H. Clowers, P. Dwivedi, W. E. Steiner, H. H. Hill Jr. and B. Bendiak, J. Am. Soc. Mass Spectrom., 2005, 16, 660–669 CrossRef CAS PubMed.
- M. D. Plasencia, D. Isailovic, S. I. Merenbloom, Y. Mechref and D. E. Clemmer, J. Am. Soc. Mass Spectrom., 2008, 19, 1706–1715 CrossRef CAS PubMed.
- P. Both, A. P. Green, C. J. Gray, R. Sardzik, J. Voglmeir, C. Fontana, M. Austeri, M. Rejzek, D. Richardson, R. A. Field, G. Widmalm, S. L. Flitsch and C. E. Eyers, Nat. Chem., 2014, 6, 65–74 CrossRef CAS PubMed.
- J. Hofmann, H. S. Hahm, P. H. Seeberger and K. Pagel, Nature, 2015, 526, 241–244 CrossRef CAS PubMed.
- A. J. Creese and H. J. Cooper, Anal. Chem., 2012, 84, 2597–2601 CrossRef CAS PubMed.
- C. Piontek, D. Varón Silva, C. Heinlein, C. Pöhner, S. Mezzato, P. Ring, A. Martin, F. X. Schmid and C. Unverzagt, Angew. Chem., Int. Ed., 2009, 48, 1941–1945 CrossRef CAS PubMed.
- K. Stavenhagen, H. Hinneburg, M. Thaysen-Andersen, L. Hartmann, D. V. Silva, J. Fuchser, S. Kaspar, E. Rapp, P. H. Seeberger and D. Kolarich, J. Mass Spectrom., 2013, 48, 627–639 CrossRef CAS PubMed.
- D. Kolarich, A. Weber, M. Pabst, J. Stadlmann, W. Teschner, H. Ehrlich, H.-P. Schwarz and F. Altmann, Proteomics, 2008, 8, 254–263 CrossRef CAS PubMed.
- R. M. Anthony, F. Nimmerjahn, D. J. Ashline, V. N. Reinhold, J. C. Paulson and J. V. Ravetch, Science, 2008, 320, 373–376 CrossRef CAS PubMed.
- D. Kolarich, A. Weber, P. L. Turecek, H.-P. Schwarz and F. Altmann, Proteomics, 2006, 6, 3369–3380 CrossRef CAS PubMed.
- M. Thaysen-Andersen, M. R. Larsen, N. H. Packer and G. Palmisano, RSC Adv., 2013, 3, 22683–22705 RSC.
- J. Nilsson, Glycoconjugate J., 2016 DOI:10.1007/s10719-016-9649-3.
- H. Sasaki, N. Ochi, A. Dell and M. Fukuda, Biochemistry, 1988, 27, 8618–8626 CrossRef CAS PubMed.
- B. Domon and C. E. Costello, Glycoconjugate J., 1988, 5, 397–409 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, additional figures. See DOI: 10.1039/c6cc01114d |
| ‡ The authors contributed equally to this work. |
| § Glycan structures are depicted using the SNFG nomenclature recommended in the Essentials of Glycobiology.3 |
| ¶ Glycan fragment annotation is based on a nomenclature introduced by Domon and Costello.29 To differentiate between peptide and oligosaccharide fragments, small and capital letters are used, respectively. |
| This journal is © The Royal Society of Chemistry 2016 |