
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploiting the equilibrium dynamics in the self-assembly of inorganic macrocycles based upon polyoxothiometalate building blocks†
Hongying
Zang
,
Andrew
Surman
,
Deliang
Long
,
Leroy
Cronin
and
Haralampos N.
Miras
*
WestCHEM, School of Chemistry, The University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: Charalampos.moiras@Glasgow.ac.uk; Tel: +44 (0)141-330-4375
First published on 16th March 2016
Abstract
We report the preparation and characterization of two new polyoxothiometalate species, {Mo16} = {(Mo2O2S2)8(OH)16(C4O4)2}4− (compound 1) and {Mo12} = {(Mo2O2S2)6(OH)12(CH3COO)2}2− (compound 2). Control of synthesis is achieved by the addition of the organic-template and further re-arrangement is controlled simply by the addition of potassium to a precursor compound yielding compound 2. The structures of the compounds are discussed and we also show that ion-mobility mass spectrometry can be used to resolve the species and their building blocks in solution.
Polyoxothiometalates1 are self-assembled chemical systems with interesting diversity arising from the ability to generate libraries of building blocks in situ and form complex structural motifs via a condensation processes.2 It has been demonstrated recently that the use of organic templates triggers the generation of kinetically controlled diverse building blocks available to assemble into thermodynamically stable complex structures under a given set of experimental variables.3 Moreover, exploration of the [Mo2O2S2]2+/C4O42− system reveals a series of available stable species giving rise to interesting structures ranging from Möbius strip {Mo10} topologies to high nuclearity nanosized clusters {Mo96} of 3.8 nm.4 Even though the available building blocks are virtually the same in each case, it was possible to direct the self-assembly towards a new equilibrium-structure and isolate new clusters with distinct structural features.
However, the extraction of crucial information regarding the underlying self-assembly processes and the inherent ability of the [Mo2O2S2]2+/C4O42− chemical system to self-establish new equilibria by a combination of thermodynamic and topological control (e.g. stability of the system vs. the structure of the templated system), is a great challenge. The main source of reliable information is offered mainly from the solid phase (primarily XRD) and the last years from solution studies utilizing high resolution electrospray ionization mass spectrometry (ESI-MS).5 For instance we have observed that the deconvolution of the spectra of such clusters is extremely challenging due to multiple species of very similar mass-to-charge ratios (m/z). This is despite the very different sizes/structures, yet still results in overlapping distribution envelopes.6,7 Thus, it is difficult to extract crucial information regarding the building block library formed in solution which could lead to better understanding of the assembly process. Ion mobility mass spectrometry (IMS-MS) has been proven an extremely useful analytical technique for the investigation of biomolecules, polymers, fullerenes,8 and less frequently in supramolecular chemistry. The resolution of multiple overlapping distribution envelopes originating from a plethora of species in the solution, in this case become possible by separating the above based on their size/shape and m/z value.
Herein, we report the preparation of two new thiometalate clusters and observation of structural transformations between the most stable building block libraries towards new species. Moreover, we present the high-resolution (HR-MS) of the isolated clusters, and demonstrate the use of IMS-MS for the resolution and characterization of polyoxothiometalate clusters for the first time. Our motivation is based on the observation that this should facilitate their continued exploration and improve the understanding of the underlying assembly processes in a similar fashion to the polyoxometalate (POM) chemistry.
The self-condensation process of the [Mo2O2S2]2+ system, depends on the pre-organization, dictated by the templating unit, and stability of the building block libraries generated from the reaction mixture.1,2 Moreover, the stability of the building blocks depends greatly upon the available coordination sites, geometry, rigidity as well as orientation of the template. For example, utilization of various carboxylate based ligands or even XO32− (X: S or Se) templates1c showed greater flexibility whilst the squarate based templates appeared to be rigid with two additional coordination sites and two different coordination modes, Fig. 1. This is potentially the reason that the generated building blocks, {(Mo2O2S2)4(OH)10(C4O4)}, {Mo8}; {(Mo2O2S2)3(OH)8(C4O4)}, {Mo6} (Fig. 1), could be used constructively under wider range of experimental conditions giving rise to a plethora of structural motifs.
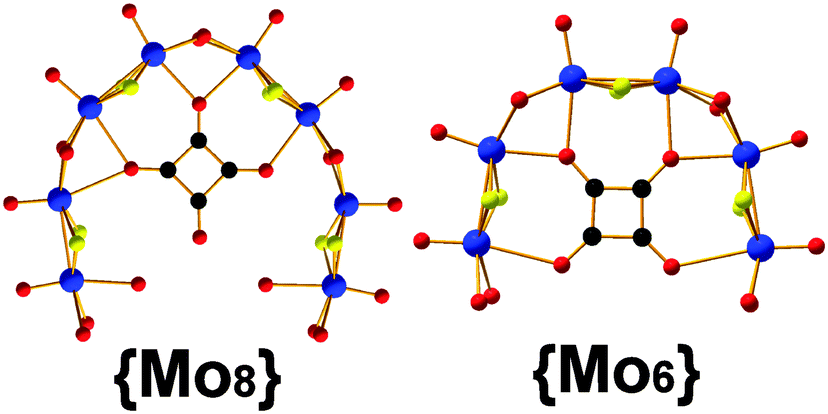 | ||
| Fig. 1 Ball-and-stick representation of the available squarate-templated building blocks {Mo8} and {Mo6}. The orientation of the squarate anion induces rigidity and steric hindrance which directs accordingly its assembly (Mo: blue; S: yellow; O: red; C: black). | ||
We have observed that the [Mo2O2S2]2+/C4O42− system is sensitive to quite small changes of reaction conditions that affect the thermodynamically stable state, by establishing a new equilibrium. Previously, we reported the template-driven assembly via (C4O4H2) of the [Mo2O2S2]2+ cations, which gave rise to the formation of a {Mo14} = (NMe4)4[(Mo2S2O2)7(OH)14(C4O4)2(H2O)2]·13H2O 3 ring-shaped cluster.4c The isolated crystalline material is possible to remain in equilibrium with the building block library formed at the early stages of the reaction for long period of times without any noticeable change. Disturbing this stable state by removing the crystalline {Mo14} product from the mother liquor, the system re-established a new equilibrium utilizing the same building block library and gave rise to a new ring-shaped {Mo16} = {(Mo2O2S2)8(OH)16(C4O4)2}4−1 cluster of increased nuclearity. In this case, the system accessed a new favourable state due to the indirect increase of the concentration of the K+ cations in the reaction mixture. In order to verify our initial claim, we followed the same experimental conditions under which the {Mo14} is normally formed in the presence of increased concentration of K+ cations, which led to the direct formation of the {Mo16} 1 species (see ESI†). Interestingly, if we dissolve crystalline pre-formed and stable in solution {Mo14} cluster in 0.5 M KCl aqueous solution at pH 5, then it is converted to the {Mo16} ring.‡ This structural transformation is quite interesting and demonstrates not only the exceptional adaptability of the system but also reflects the kinetic stability of the {Mo6} and {Mo8} building blocks (Fig. 1), introduced by the C4O42− template.
Compound 1 crystallises in the C2/m space group exhibiting an idealized D2h symmetry. The cluster is constructed by eight [Mo2O2S2]2+ units bridged by two hydro-oxo groups and templated by two squarate anions forming an ellipsoid architecture. Interestingly in this case the {Mo16} consists of two {Mo6} building blocks (Fig. 1) in contrast to the {Mo14} ring. Additionally, there is incorporation of two [Mo2O2S2]2+ cations between the {Mo6} building units which are responsible for the observed ring expansion. Moreover, a K+ cation is captured within the cavity (K–O = 2.82(3), 2.87(4) Å) between the two squarate templates inducing further structural stability to the oxothiometalate framework.
In an effort to generate a more flexible building block library, we used acetate anions as template for the self-condensation of [Mo2O2S2]2+. In this case a dodecamolybdate ring {Mo12} = {(Mo2O2S2)6(OH)12(CH3COO)2}·24H2O 2 crystallized within two weeks (Fig. 2). The compound was synthesized from a [Mo2O2S2]2+ aqueous medium in the presence of CH3COONa at pH 4.5. X-ray diffraction analysis revealed an inorganic cyclic neutral skeleton {Mo2nS2nO2n(OH)2n} (n = 6), which incorporates two acetate anionic templates. The wheel is formed by six [Mo2O2S2]2+ building blocks connected to each other through double hydroxo bridges. Two types of Mo–Mo bonds are observed: a short Mo–Mo distance found to be ca. 2.8 Å within the dinuclear unit, characteristic of a metal–metal bond, and the long Mo–Mo distance which found to be ca. 3.4 Å which are in agreement with the literature.9 The molecular arrangement of the wheel exhibits a 12-membered ring encapsulating two acetates residing parallel to the plane of the ring. The incorporation of the acetate template introduces distortion to the {Mo12} ring which lowers its idealized symmetry to C2h.9 Two coordination water molecules attach onto two MoV atoms (Mo–Ow = 2.42(6) Å) and possibly due to the steric hindrance introduced by the by –CH3 groups of the acetate templates, four MoV atoms found to be five-coordinated. In an effort to verify the kinetic stability of the generated building block library, we disturbed the existing equilibrium by increasing the concentration of K+/or adding Cs+ cations and let the system access an alternative stable thermodynamic state. In this case no transformation has been observed. This is potentially due to the fact that the flexibility of the acetate templates induce structural instability of the respective building block in marked contrast to the squarate based ones, Fig. 1.
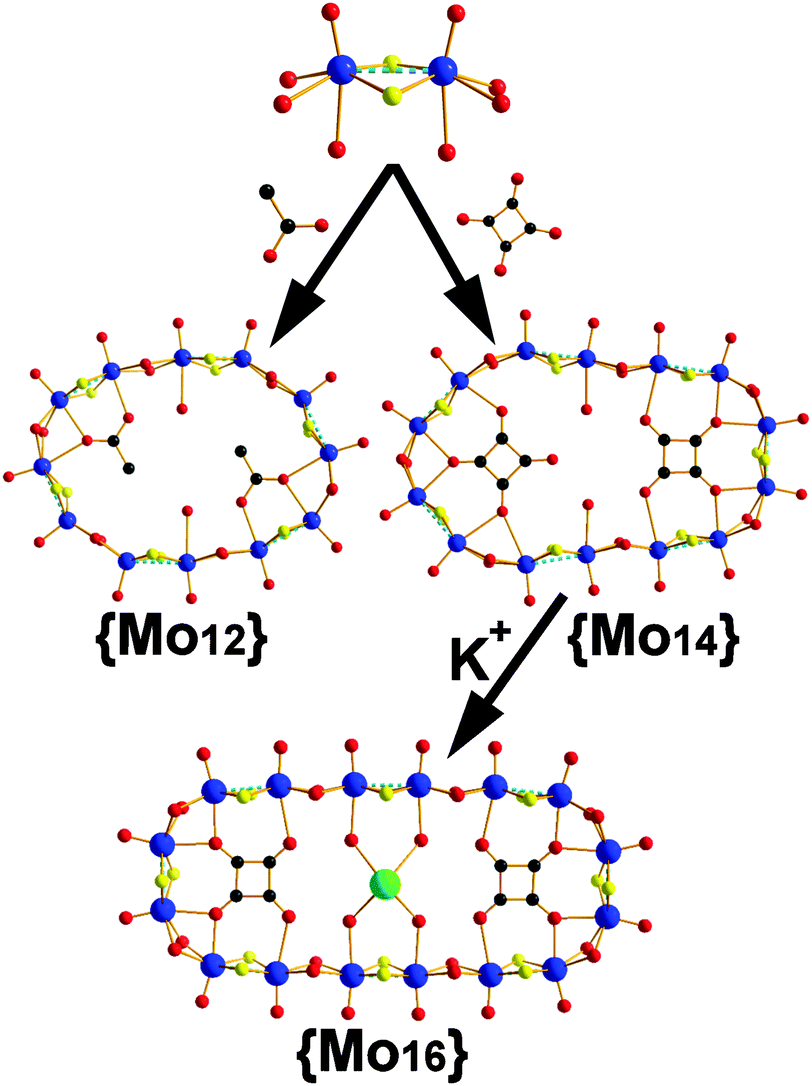 | ||
| Fig. 2 Templated directed assembly of [Mo2O2S2]2+ dimeric units leading to 1 {Mo12} and {Mo14} which is transformed further to 2 {Mo16} ring associated with a 45° degree template rotation (Mo: blue; S: yellow; O: red; K: green; C: black). | ||
High resolution negative mode ESI-MS was used in conjunction with the structural analysis to confirm the identity of clusters 1 and 2. Most of the major peaks observed Fig. S7 (ESI†) are broad and plausibly assigned as a continua resulting from a single anion with a range of different cations and/or neutral addenda (particularly solvent ions); this is characteristic of such compounds in general and a very frequent problem in polyoxothiometalate chemistry. Thus, Travelling Wave Ion Mobility MS (TWIMS) was used here to resolve overlapping peak sets. IMS-MS (Fig. 3 and Fig. S8, ESI†) and CID studies (vide infra) (Fig. S9, ESI†) support the assignment of the broad peaks ca. 1000 Da (z = −3) and 1500 Da (z = −2) as corresponding to the intact {Mo16} cluster with a variety of cations/solvent molecules (listed in blue fonts) (Table S1, ESI†), and most other peaks are assigned as fragments (some of the largest listed in red fonts). Drift time through an N2-filled drift tube is plotted against m/z, with intensity represented in colour shading at each point (here in a linear scale); peaks corresponding to the same (or similarly-sized) ions fall on a shallow parabolic curve or line.10 The recorded spectrum revealed peaks falling into three sets – one consistent with intact {Mo16} clusters (marked in blue), and two consistent with smaller fragments (marked in red), Fig. S8 (ESI†). Interestingly, IMS-MS reveals two conformations in the −2-charged ion at ca. 1500 Da; this may correspond to different arrangements of the bound water and counter-cations. Furthermore, a very faint peak can be observed on the same vertical line as there two large peaks (z = 4), corresponding to an ion incorporating two such clusters; such dimers are commonly observed in the gas phase polyoxometalate clusters.10
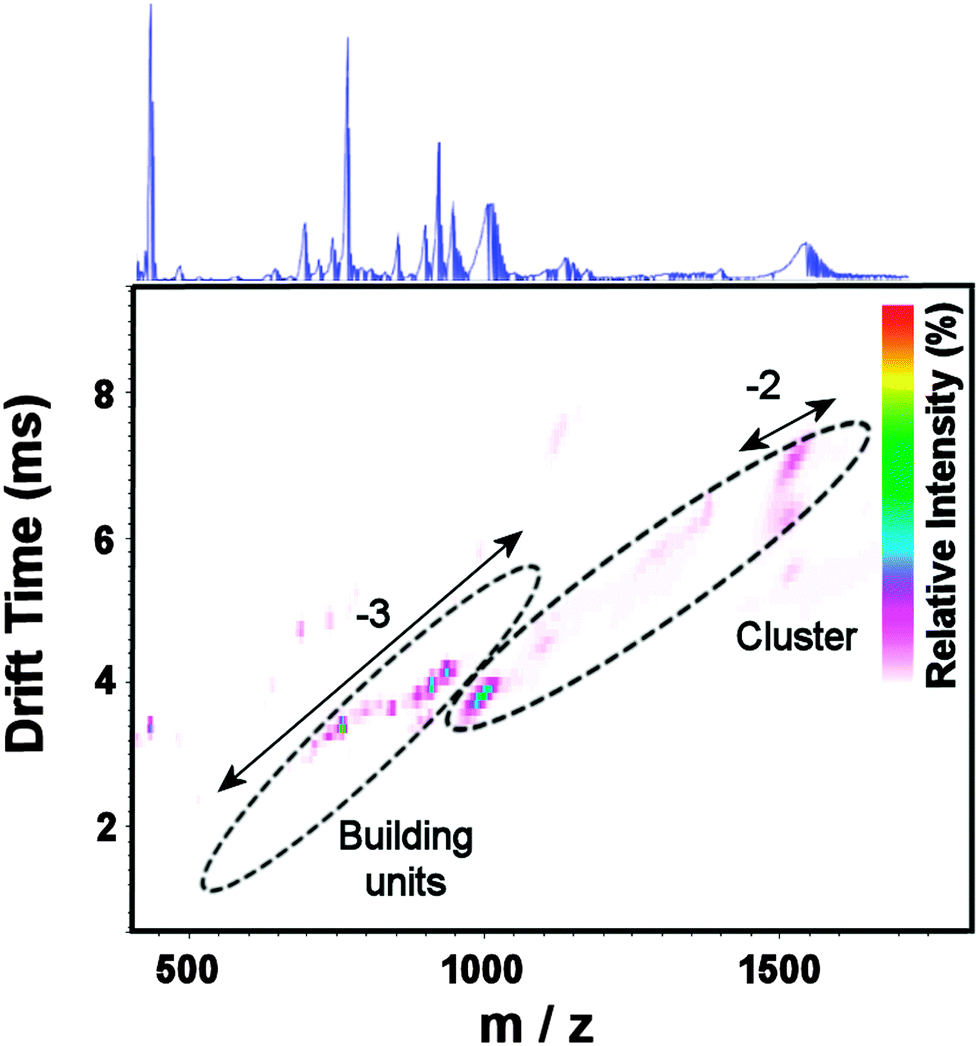 | ||
| Fig. 3 ESI-IMS-MS of {Mo16} 1 in MeOH. Relative intensity (cf. highest peak) is shown on a linear scale (coloured scale, inset); samples of isotope patterns (inset) were obtained in resolution mode; see ESI† for details. | ||
Interestingly we were able to observe the existence of the intact cluster and the derivatives of the {Mo6} building block after deconvolution of the MS spectrum unambiguously. By contrast, compound 2 proved to be extremely fragile under ESI-MS conditions (Fig. S10, ESI†): that we were not able to observe neither the intact {Mo12} 2 cluster, nor its acetate templated building block derivatives, is an indication of its instability.
In summary, we have discussed the preparation of the two new polyoxothiometalates species by generating two building block libraries of different stability which led to the formation of hexadecanuclear, {(Mo2O2S2)8(OH)16(C4O4)2}4−1, and a dodecanuclear, {(Mo2O2S2)6(OH)12(CH3COO)2}2−2, ring constructed by squarate and acetate templated building blocks respectively. Moreover, we observed experimentally an intriguing system showing a type of template-based adaptability via the interplay of the [Mo2O2S2]2+/C4O42− units where a structural transformation of {Mo14} to {Mo16} took place utilizing different available constituents of the building block library. The transformation was triggered by the addition of potassium cations forcing the system to access a new thermodynamic minimum. Finally, we demonstrated the efficacy of the IMS-MS technique for the first time in polyoxothiometalate chemistry, which helped us not only characterize the {Mo16} ring in solution, but also to identify stable derivatives of the building blocks used for its construction.
The authors would like to thank the EPSRC grants (No. EP/J015156/1; EP/L023652/1; EP/I033459/1; EP/J015156/1; EP/K023004/1; EP/L023652/1), EC grant 318671 MICREAGENTS, LC thanks the Royal Society/Wolfson Foundation for a Merit Award and the ERC for an Advanced Grant (ERC-ADG, 670467 SMART-POM).
Notes and references
-
(a) E. Cadot, M. N. Sokolov, V. P. Fedin, C. Simonnet-Jegat, S. Floquet and F. Secheresse, Chem. Soc. Rev., 2012, 41, 7335–7353 RSC
; (b) S. Duval, S. Floquet, C. Simonnet-Jégat, J. Marrot, R. N. Biboum, B. Keita, L. Nadjo, M. Haouas, F. Taulelle and E. Cadot, J. Am. Chem. Soc., 2010, 132, 2069–2077 CrossRef CAS PubMed
-
(a) U. Kortz, S. S. Hamzeh and N. A. Nasser, Chem. – Eur. J., 2003, 9, 2945–2952 CrossRef CAS
-
(a) J.-F. Lemonnier, S. Floquet, J. Marrot, E. Terazzi, C. Piguet, P. Lesot, A. Pinto and E. Cadot, Chem. – Eur. J., 2007, 13, 3548–3557 CrossRef CAS PubMed
-
(a) H. Y. Zang, H. N. Miras, J. Yan, D.-L. Long and L. Cronin, J. Am. Chem. Soc., 2012, 134, 11376–11379 CrossRef CAS PubMed
- H. N. Miras, D.-L. Long, P. Kögerler and L. Cronin, Dalton Trans., 2008, 214–221 RSC
- H. N. Miras, J. Yan, D. L. Long and L. Cronin, Angew. Chem., Int. Ed., 2008, 47, 8420–8423 CrossRef CAS PubMed
- H. N. Miras, H. Y. Zang, D. L. Long and L. Cronin, Eur. J. Inorg. Chem., 2011, 5105–5111 CrossRef CAS
-
(a) D. E. Clemmer and M. F. Jarrold, J. Mass Spectrom., 1997, 32, 577–592 CrossRef CAS
- E. Cadot, B. Salignac, S. Halut and F. Sécheresse, Angew. Chem., Int. Ed., 1999, 37, 611–612 CrossRef
- P. J. Robbins, A. J. Surman, J. Thiel, D.-L. Long and L. Cronin, Chem. Commun., 2013, 49, 1909–1911 RSC
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, structure figures, IR, UV-vis, TGA, MS experimental details and peak assignment. CCDC 1451290(1) and 1451291(2). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc01041e |
| ‡ Crystallographic data for compound 1: C12H96K3Mo16NO74S16, Mr = 3604.19 g mol−1; yellow block crystal; monoclinic, space group C2/m, a = 20.6782(10), b = 11.2433(5), c = 23.3822(11), V = 5151.2(4) Å3, Z = 2. Crystallographic data for compound 2: C12H90Mo12N2O52S12, Mr = 2630.85 g mol−1; yellow block crystal; monoclinic, space group C2/c, a = 38.310(2), b = 17.3787(9), c = 12.5611(7), V = 8360.2(8) Å3, Z = 4. CCDC 1451290(1) and 1451291(2). |
| This journal is © The Royal Society of Chemistry 2016 |