
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective preparation of elusive and alternative single component polymorphic solid forms through multi-component crystallisation routes†
Lynne H.
Thomas
a,
Craig
Wales
ab and
Chick C.
Wilson
*ac
aDepartment of Chemistry, University of Bath, Bath BA2 7AY, UK. E-mail: C.C.Wilson@bath.ac.uk
bSchool of Chemistry, University of Glasgow, Glasgow G12 8QQ, UK
cCMAC, EPSRC Centre for Continuous Manufacturing and Crystallisation, Department of Chemistry, University of Bath, Bath BA2 7AY, UK
First published on 6th April 2016
Abstract
A transferable, simple, method for producing previously elusive and novel polymorphic forms of important active pharmaceutical ingredients (APIs; paracetamol (acetaminophen), piroxicam and piracetam) is demonstrated. Nitrogen heterocyclic co-molecules are employed to influence the self-assembly crystallisation process in a multi-component environment. Previously unknown solvates have also been synthesised by this method.
Polymorph control is vital in the design and formation of many solid state materials. Different solid forms of a material can exhibit very different physical properties, including solubility, bioavailability, compressibility and thermal stability. The phenomenon attracts a high level of interest both for fundamental research and in applications, particularly in domains such as the pharmaceutical industry. One primary application of polymorphism has, therefore, been focused towards the formulation of solid crystalline forms of active pharmaceutical ingredients (APIs),1–3 as the potential exists for altering physical properties that can enhance delivery and bioavailability without compromising molecular form and biological activity of an API.4 This tuning of properties by polymorph selection and control is not restricted to APIs, with applications in a wide variety of functional materials including porous materials with potential gas storage applications,5,6 agrochemicals,7 pigments and dyes,8 explosives9 and non-linear optical materials (NLOs).10 Reliable, transferable routes for directing the formation of a metastable polymorph of any type have previously been elusive, with complex routes including the use of temperature cycling (tempering), additives, polymers or high pressure often being the only option.
The assembly of molecular materials into solid forms is frequently achieved via crystallisation from solution, often in an evaporative environment. Crystallisation conditions are routinely varied to optimise the conditions under which polymorphs can be isolated. In particular, additives have been used to direct polymorph formation; these are normally used in trace quantities11,12 or are engineered to have a structural relationship between the additive and the desired polymorph, thus facilitating epitaxial growth on the substrate surface13 or, conversely, inhibiting the growth of the more stable form.14
We have previously reported15 the production of the elusive and pharmaceutically relevant second polymorph of p-hydroxyacetanilide (paracetamol/acetaminophen), in 100% yield through multi-component evaporative crystallisation, utilising as co-components a range of mono-substituted benzoic acids and other compounds containing carboxylic acid groups. Importantly, the outcome from this method differs from the conventional co-crystallization method, as the benzoic acid does not complex with paracetamol, instead crystallising independently, whilst perturbing the solution environment to direct crystallisation towards the metastable paracetamol form II rather than the thermodynamically more favourable form I. The method offers 100% yields of form II and unprecedented simplicity compared with previous routes to this form. In contrast to conventional additive crystallisations, the co-crystallisation approach adopted here uses molar quantities of both the active component and of the benign templating molecule. Co-crystallisation from multi-component environments is relatively rare as a route to individual polymorph selection, being more conventionally applied to molecular complex (co-crystal) formation where the physical properties are altered by introducing a second component into the resulting solid form.16 Discoveries of new and elusive single component polymorphs from co-crystallisation experiments have until now been serendipitous outcomes from such experiments,17–19 including with N-heterocycles.20
Here we report the establishment of a systematic co-crystallisation methodology utilising nitrogen heterocycles as an intentional, designed, route directing the self-assembly of new, elusive and novel crystal forms of various single component materials (Table 1), namely the non-steroidal anti-inflammatory drug (NSAID) piroxicam (1), the nootropic pharmaceutical piracetam (3) and the trihydroxybenzoic acid gallic acid (4; which also has pharmaceutical properties21) as well as identifying further multicomponent routes to the analgesic paracetamol (2) form II. We highlight the diverse types of crystal forms accessible using this method, showing that the combination of co-crystallisation of the active ingredients with nitrogen-heterocycles present in excess, along with elevated temperatures of crystallisation, repeatedly directs the formation of a range of both elusive and previously unknown crystal forms. The use of low melting point solids allows these to evaporate along with the solvent leaving only the target component present in the solid product, an extremely favourable experimental outcome in terms of isolation of the desired solid form.
| 1-III | 1-IV | 1-MeOH | 4-H2O | |
|---|---|---|---|---|
| a | 7.8557(3) | 12.6899(9) | 7.037(2) | 12.554(2) |
| b | 10.1043(4) | 14.6928(9) | 12.917(4) | 8.8499(15) |
| c | 10.4713(5) | 20.5097(16) | 25.467(8) | 12.6253(17) |
| α | 80.602(2) | 84.866(2) | 84.077(7) | 90 |
| β | 68.952(2) | 74.669(3) | 89.174(7) | 103.988(5) |
| γ | 69.742(2) | 84.683(2) | 83.327(6) | 90 |
| Space grp |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
P21/n |
Piroxicam (1) has been studied extensively in the solid state,22 but the literature on piroxicam polymorphism is largely conflicting in terms of the number and nomenclature of polymorphs. This was clarified by Sheth,23 establishing that there are three known polymorphs, including form III, for which no crystal data have yet been reported; a monohydrate is also known.24 Recently, a further polymorph has been suggested, formed using electrospray methods, although the particle sizes were small and it was not possible to determine the crystal structure.25 Further evidence for this form is required. Piracetam form I is another elusive polymorphic form which has been difficult to isolate and has previously only been produced by quenching of the form III polymorph.26 No evaporative route to piracetam form I has previously been reported. There is a known anhydrous form of gallic acid crystallised from methanol/chloroform solvent,27 and four known polymorphs of the monohydrate.21c In the latter, forms I and II crystallise in monoclinic space groups P21/c and P2/n, respectively, form III is a high Z′ triclinic form obtained via attempted co-crystallisation of gallic acid and sarcosine in aqueous methanol, while monoclinic form IV results from aqueous crystallisation.
The most versatile co-molecule/temperature regime found for these templating routes is that using pyrazole at 50 °C (Fig. 1).‡ The elusive polymorphs piroxicam form III (1-III; acetonitrile), piracetam form I (1-I; methanol) and paracetamol form II (2-II; methanol) could be isolated using solvent evaporation in the presence of pyrazole. Piroxicam form III (1-III) could also be templated using the related 3,5-dimethylpyrazole in otherwise identical crystallisation conditions. Extending the class of nitrogen heterocycle used to pyrazine, crystallisation in the multi-component environment, again at 50 °C, leads to the selective growth of paracetamol form II (2-II; methanol, 100% yields) and a new polymorph of piroxicam, denoted form IV (1-IV; acetonitrile; this can also be produced at 40 °C, although less reliably).§
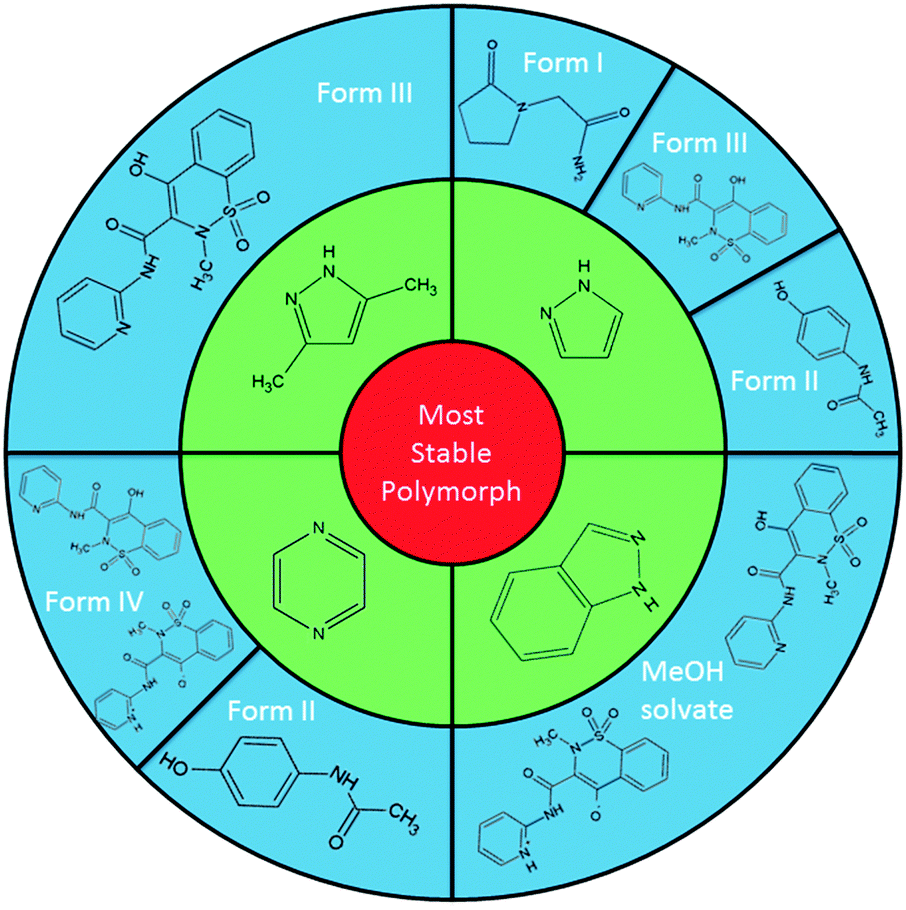 | ||
| Fig. 1 Multi-component routes to elusive polymorphs of piroxicam, piracetam and paracetamol. In this schematic, the green ring represents the various N-heterocycles employed, the outer blue ring the target molecules, detailing the forms produced by the templating approach. | ||
Piroxicam form III (1-III) has therefore been successfully isolated and its full single crystal structure determined for the first time, confirmed by a good match to the powder X-ray diffraction (PXRD) pattern for the phase identified as piroxicam form III reported previously [see S2.1.1, ESI†].22d,23 The crystal structure of 1-III consists of a single molecule in the asymmetric unit which forms the same piroxicam dimers found in piroxicam form I,22avia N–H⋯O hydrogen bonds between the amide N–H and the sulfonyl oxygen atoms. The structure of form III is also layered in a similar fashion to form I (Fig. 2), with the methyl and sulfonyl groups bridging between the layers. The differences between form I and form III are seen in the extended packing and the weaker hydrogen bonds, detailed in the ESI.†
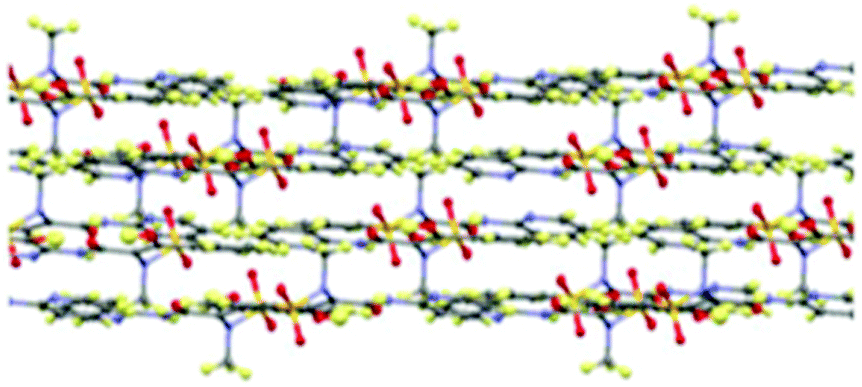 | ||
| Fig. 2 The layered arrangement of molecule in the crystal packing in piroxicam form III, 1-III. | ||
Previous routes to this form have involved far more complex procedures, including cryo-grinding to obtain an amorphous phase followed by heating23 or spraying a saturated ethanol solution on dry ice22d to produce a form III powder. The ability to produce significant product volumes as well as good quality crystals suitable for single crystal X-ray diffraction from simple evaporative routes, as reported here, offers obvious advantages over these techniques. Form I, II or the monohydrate are often co-existent with form III, the latter being isolable as it is present as large block-shaped crystals. The new solid form of piroxicam reported here, form IV (1-IV), is unique as it is the only known polymorph in which piroxicam molecules are present in both the non-ionised and zwitterionic tautomers (see ESI,† S2.2); further, this form has a surprisingly large asymmetric unit, containing four neutral and one zwitterionic molecules. Prior to our discovery of this fourth polymorph with mixed tautomeric states, pure piroxicam in the solid form was only known to exist as the non-ionised tautomer, with the zwitterionic form only found in the monohydrate24 and other multi-component complexes.28,29 There is a substantial conformational difference between the neutral and zwitterionic forms, providing clear evidence of the assignment in addition to the different distribution of hydrogen atoms within the tautomers. The neutral piroxicam molecules dimerise through two N–H⋯N hydrogen bonds (hydrogen bonding is fully described in the ESI†), with two symmetry independent dimers formed in this way. The hydrogen bonding in this dimer is unique from that seen in any of the other three polymorphs of piroxicam. Equivalent zwitterionic piroxicam molecules also dimerise, although the presence of the different tautomer results in a completely different type of dimer to that formed by the neutral molecules. This type of dimer is formed by zwitterionic molecules in all previously reported zwitterionic molecular complexes.24,28 Yields approaching 100% of form IV could be obtained, resulting in small needle-shaped crystals suitable for full structural characterisation.
In extensions of this multi-component approach, we have successfully discovered new forms of these and other common APIs. For example, a methanol solvate of piroxicam (1-MeOH) was crystallised using indazole as a co-molecule and a recrystallisation temperature of 60 °C (see ESI,† S2.3); this solvate also features a mixture of both neutral and zwitterionic tautomers (two zwitterionic to one neutral). This is only the second piroxicam complex to feature co-existence of both tautomers of piroxicam in the same crystal structure.281-MeOH crystallises in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio. The symmetry independent zwitterionic molecules dimerise as in 1-IV, via two moderately strong charge-assisted bifurcated N+–H⋯O hydrogen bonds (hydrogen bonding is fully described in the ESI†). The MeOH molecule lies below the plane of the dimer and links to one of the zwitterionic molecules through a further moderately strong, charge assisted O–H⋯O− hydrogen bond. The overall structure is layered (Fig. 3) with the MeOH molecule linking the neutral molecules to zwitterionic molecules above. Co-crystallisation of piracetam (3) with gallic acid (4; GA) produces a new polymorph of gallic acid monohydrate (porous-GAM, 4-H2O; see ESI,† S3.1). This new polymorph is porous, with large channels containing disordered solvent running through the crystal structure (Fig. 4). There is a single GA and a single water molecule in the asymmetric unit. The GA molecules form a typical carboxylic acid dimer unit via moderately strong O–H⋯O hydrogen bonds (Fig. S3.1.1, ESI†). Weaker O–H⋯O hydrogen bonds between the hydroxyl groups (Fig. S3.1.1, ESI†) link the GA molecules into large rings forming the structural frame of the channels. Further hydrogen bonding involves the water molecule resolved in the structure (see ESI†). The channels are a significant proportion of the unit cell volume (Fig. 4), with approximately 5.0% of the unit cell volume (47.60 Å3) accessible to solvent water molecules (using a probe of van der Waals radius 1.4 Å). The channels contain significant, unresolved, disordered solvent molecules. 4-H2O is isolated from room temperature multi-component crystallisation from methanol, ethanol, acetonitrile or acetone solution containing an approximate 1:1 molar ratio of GA (from a mixture of the anhydrous form and GA monohydrate form II) and piracetam (form III). The large pore volume in the solid form of porous-GAM (4-H2O) is of clear interest and the gas storage potential17,30 of this material is under investigation. To demonstrate the subtle balance that can occur in these multi-component preparations, recrystallisation from ethyl acetate did not produce the porous GA solvate, instead resulting in the formation of a 1:1 molecular complex between piracetam and GA (3–4; see ESI,† S3.2).
:1 molar ratio. The symmetry independent zwitterionic molecules dimerise as in 1-IV, via two moderately strong charge-assisted bifurcated N+–H⋯O hydrogen bonds (hydrogen bonding is fully described in the ESI†). The MeOH molecule lies below the plane of the dimer and links to one of the zwitterionic molecules through a further moderately strong, charge assisted O–H⋯O− hydrogen bond. The overall structure is layered (Fig. 3) with the MeOH molecule linking the neutral molecules to zwitterionic molecules above. Co-crystallisation of piracetam (3) with gallic acid (4; GA) produces a new polymorph of gallic acid monohydrate (porous-GAM, 4-H2O; see ESI,† S3.1). This new polymorph is porous, with large channels containing disordered solvent running through the crystal structure (Fig. 4). There is a single GA and a single water molecule in the asymmetric unit. The GA molecules form a typical carboxylic acid dimer unit via moderately strong O–H⋯O hydrogen bonds (Fig. S3.1.1, ESI†). Weaker O–H⋯O hydrogen bonds between the hydroxyl groups (Fig. S3.1.1, ESI†) link the GA molecules into large rings forming the structural frame of the channels. Further hydrogen bonding involves the water molecule resolved in the structure (see ESI†). The channels are a significant proportion of the unit cell volume (Fig. 4), with approximately 5.0% of the unit cell volume (47.60 Å3) accessible to solvent water molecules (using a probe of van der Waals radius 1.4 Å). The channels contain significant, unresolved, disordered solvent molecules. 4-H2O is isolated from room temperature multi-component crystallisation from methanol, ethanol, acetonitrile or acetone solution containing an approximate 1:1 molar ratio of GA (from a mixture of the anhydrous form and GA monohydrate form II) and piracetam (form III). The large pore volume in the solid form of porous-GAM (4-H2O) is of clear interest and the gas storage potential17,30 of this material is under investigation. To demonstrate the subtle balance that can occur in these multi-component preparations, recrystallisation from ethyl acetate did not produce the porous GA solvate, instead resulting in the formation of a 1:1 molecular complex between piracetam and GA (3–4; see ESI,† S3.2).
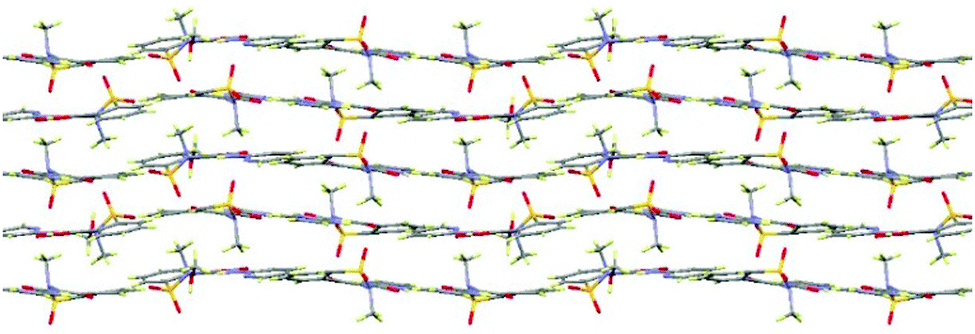 | ||
| Fig. 3 Layers in the piroxicam: methanol (3:1) solvate, I-MeOH. | ||
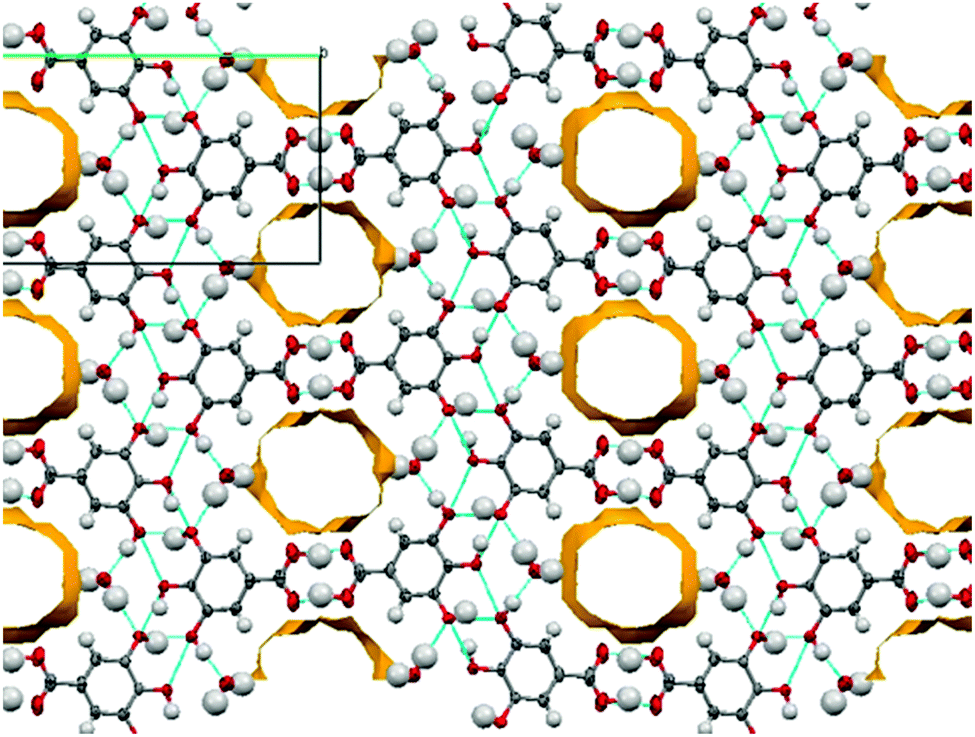 | ||
| Fig. 4 Channels observed in the porous form of gallic acid monohydrate (4-H2O). | ||
The mechanism by which certain co-molecules direct the formation of a particular crystal form of another material in these multi-component crystallisations remains unclear. The evidence in the present study suggests a solution-based mechanism within the multi-component environment, rather than epitaxy; no intergrowths of crystals with the co-molecule were observed and, in the case of pyrazole and pyrazine where the melting points are similar to the temperature of crystallisation, the co-molecule was not present in the crystallisation vessel on complete evaporation of the solvent. Extensive control experiments without the co-molecules present in the crystallisation environment showed no sign of the elusive polymorphs, from screens using single-crystal or powder XRD, while variation of the solvent or temperature resulted in the recrystallisation of the known stable forms. The variety of important solid form materials found to be templated using N-heterocycles in the present work makes this a viable alternative strategy towards polymorph selection, by applying multicomponent crystallisation in a targeted rather than serendipitous approach.
In summary, we have shown that solid form assembly using multi-component crystallisation conditions where mass or molar ratios of the benign co-component are utilised along with an elevated temperature of crystallisation can lead to new forms of various APIs. The range of crystal forms obtainable by this method includes the production of both previously elusive and novel solid forms of well-studied APIs, along with the generation of single phases containing mixed tautomeric forms of piroxicam (1-IV) and a porous polymorph of gallic acid monohydrate (4-H2O). We have established this method as a versatile and widely applicable alternative methodology for polymorph control. The range of nitrogen heterocycles which show the ability to direct the crystallisation towards the elusive polymorphs, paracetamol form II (2-II) and piroxicam forms III (1-III) and IV (1-IV), delivers a systematic and directed approach. Otherwise difficult to obtain polymorphs can by this means be crystallised in significant quantities from simple evaporation at elevated temperatures of crystallisation. The multi-component enabled crystallisation of piroxicam forms III and IV is more sensitive to the crystallisation environment than that of paracetamol form II, possibly due to the increased competition incurred by the existence of the more stable forms I and II as well as the monohydrate. Details of the relative stabilities of these four polymorphs will be reported elsewhere. The evidence from these experiments indicates that the mechanism is solution-based rather than epitaxial, as proposed in our previous work with paracetamol form II (2-II)15 but the use of nitrogen heterocycles also requires the use of elevated temperatures of crystallisation to enable selection of the metastable polymorphic forms.
This work was supported by the Scottish Funding Council SPIRIT programme and the EPSRC Centre for Innovative Manufacturing in Continuous Manufacturing and Crystallisation (CMAC; EP/I033459/1).
Notes and references
- J. Haleblian and W. McCrone, J. Pharm. Sci., 1969, 8, 91 Search PubMed.
- G. P. Stahly, Cryst. Growth Des., 2007, 7, 1007 CAS.
- J. Bauer, S. Spanton, R. Henry, J. Quick, W. Dziki, W. Porter and J. Morris, Pharm. Res., 2001, 18, 859 CrossRef CAS.
- N. Schultheiss and A. Newman, Cryst. Growth Des., 2009, 9, 2950 CAS.
- S. V. Lindeman, V. E. Shklover and Y. T. Struchkov, Cryst. Struct. Commun., 1981, 10, 1173 CAS.
- J. L. Daschbach, T. Chang, L. R. Corrales, L. X. Dang and P. McGrail, J. Phys. Chem. B, 2006, 110, 17291 CrossRef CAS PubMed.
- D. Chopra, T. P. Mohan, K. S. Rao and T. N. G. Row, CrystEngComm, 2005, 7, 374 RSC.
- P. Erk, H. Hengelsberg, M. F. Haddow and R. van Gelder, CrystEngComm, 2004, 6, 474 RSC.
- D. I. A. Millar, I. D. H. Oswald, D. J. Francis, W. G. Marshall, C. R. Pulham and A. S. Cumming, Chem. Commun., 2009, 562 RSC.
- Y. Wang, W. Tam, S. H. Stevenson, R. A. Clement and J. Calabrese, Chem. Phys. Lett., 1988, 148, 136 CrossRef CAS.
- A. D. Bond, K. A. Solanko, S. Parsons, S. Redder and R. Boese, CrystEngComm, 2011, 13, 399 RSC.
- N. K. Nath and A. Nangia, CrystEngComm, 2011, 13, 47 RSC.
- K. Chadwick, A. Myerson and B. Trout, CrystEngComm, 2011, 13, 6626 RSC.
- R. J. Davey and N. Blagden, J. Am. Chem. Soc., 1997, 119, 1767 CrossRef CAS.
- L. H. Thomas, C. Wales, L. Zhao and C. C. Wilson, Cryst. Growth Des., 2011, 11, 1450 CAS.
- A. S. Cannon and J. C. Warner, Cryst. Growth Des., 2002, 2, 255 CAS.
- N. K. Nath, S. S. Kumar and A. Nangia, Cryst. Growth Des., 2011, 11, 4594 CAS.
- M. Rafilovich and J. Bernstein, J. Am. Chem. Soc., 2006, 128, 12185 CrossRef CAS PubMed.
- G. M. Day, A. V. Trask, W. D. S. Motherwell and W. Jones, Chem. Commun., 2006, 54 RSC.
- P. Sanphui, N. R. Goud, U. B. R. Khandavilli, S. Bhanoth and A. Nangia, Chem. Commun., 2011, 47, 5013 RSC.
- (a) Y. Lu, F. Jiang, H. Jiang, K. Wu, X. Zheng, Y. Cai, M. Katakowski, M. Chopp and S. S. T. To, Eur. J. Pharmacol., 2010, 641, 102 CrossRef CAS PubMed; (b) D. H. Priscilla and P. S. M. Prince, Chem.-Biol. Interact., 2009, 179, 118 CrossRef CAS PubMed; (c) H. D. Clarke, K. K. Arora, Ł. Wojtas and M. J. Zaworotko, Cryst. Growth Des., 2011, 11, 964 CrossRef CAS; (d) X. Zhao, W. Zhang and S. J. Kong, J. Liq. Chromatogr. Relat. Technol., 2007, 30, 235 CrossRef CAS.
- (a) B. Kojic-Prodic and Z. Ruzic-Toros, Acta Crystallogr., 1982, B38, 2948 CrossRef CAS; (b) F. Vrecer, S. Srcic and J. Smid-Korbar, Int. J. Pharm., 1991, 68, 35 CrossRef CAS; (c) A. R. Sheth, S. Bates, F. X. Muller and D. J. W. Grant, Cryst. Growth Des., 2005, 5, 571 CrossRef CAS; (d) F. Vrecer, M. Vrbinc and A. Meden, Int. J. Pharm., 2003, 256, 3 CrossRef CAS PubMed.
- A. R. Sheth, S. Bates, F. X. Muller and D. J. W. Grant, Cryst. Growth Des., 2004, 4, 1091 CAS.
- J. Bordner, J. A. Richards, P. Weeks and E. B. Whipple, Acta Crystallogr., 1984, C40, 989 CAS.
- M. Nyström, J. Roine, M. Murtomaa, R. M. Sankaran, H. A. Santos and J. Salonen, Eur. J. Pharm. Biopharm., 2015, 89, 182 CrossRef PubMed.
- D. Louer and M. Louer, Acta Crystallogr., 1995, B51, 182 CAS.
- J. Zhao, I. A. Khana and F. R. Fronczek, Acta Crystallogr., 2011, E67, o316 Search PubMed.
- S. L. Childs and K. I. Hardcastle, Cryst. Growth Des., 2007, 7, 1291 CAS.
- M. Inoue, R. Suzuki, N. Sakaguchi, Z. Li, T. Takeda, Y. Ogihara, B. Y. Jiang and Y. Chen, Biol. Pharm. Bull., 1995, 18, 1526 CAS.
- J. R. Holst, A. Trewin and A. I. Cooper, Nat. Chem., 2010, 2, 915 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallisation conditions, PXRD confirming phase yield of 2 form-II, identity of 1-IIIcf. previous literature, production of 3-I, single crystal structure determination details and structural descriptions for 1-III, 1-IV, 1-MeOH, 4-H2O & 3–4. CCDC 1451204–1451208. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc01027j |
| ‡ Full details of molar ratios and other crystallisation conditions attempted in the multi-component crystallisations reported are given as ESI.† |
| § The focus here is on solid form discovery; the single crystals isolated unambiguously establish the production of novel solid forms, but are not necessarily the exclusive or majority product. |
| This journal is © The Royal Society of Chemistry 2016 |