
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aurophilicity under pressure: a combined crystallographic and in situ spectroscopic study†
Alice E.
O'Connor
a,
Nedaossadat
Mirzadeh
*ab,
Suresh K.
Bhargava
b,
Timothy L.
Easun
 c,
Martin
Schröder
*ad and
Alexander J.
Blake
*a
c,
Martin
Schröder
*ad and
Alexander J.
Blake
*a
aSchool of Chemistry, The University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: a.j.blake@nottingham.ac.uk
bCentre for Advanced Materials & Industrial Chemistry, School of Applied Sciences, RMIT University, GPO Box 2476V, Melbourne, Victoria 3001, Australia
cSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK
dSchool of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
First published on 11th March 2016
Abstract
High pressure crystallographic studies on [1,4-C6H4{PPh2(AuCl)}2] (1) reveal the largest pressure-induced contraction of an aurophilic interaction observed for any Au(I) complex; Hirshfeld surface analysis and Raman spectroscopy reveal the presence of several types of intermolecular interaction, which play an important role in the behaviour of 1 as a function of pressure.
The term aurophilicity refers to the tendency of Au complexes to aggregate via the formation of weak Au–Au bonds.1 The aurophilic interaction is comparable in strength to moderate hydrogen bonding, making it of particular interest in the formation of supramolecular structures held together by relatively weak interactions.2 The binding energy of aurophilic interactions is 20–60 kJ mol−1,3 whilst π⋯π interactions are considerably weaker. Typical Au⋯Au interatomic distances for Au(I) complexes, elemental gold and gold clusters fall in the range 2.5–3.2 Å and are shorter than the sum of the van der Waals radii for two gold atoms (3.32 Å).4
Although evidence for the phenomenon of aurophilicity is derived principally from the wealth of knowledge provided by crystal structure analysis, the nature of the Au–Au interaction has been the subject of many pioneering computational studies. Aurophilicity may be described as a correlation effect enhanced by relativistic effects.5–7 The correlation contribution of the binding energy has been predicted using local second-order Møller–Plesset perturbation theory (LMP2) in model dimers (A–B) of type [X–Au–PH3]2 (X = H, Cl), revealing the equal contribution of van der Waals (A → A′, B → B′) and ionic excitations (A → A′, B → A′).8 Interestingly, extended calculations at the CCSD(T) level and dispersion-corrected density-functional theory have emphasized the role of the method of calculation, and suggest that relativistic effects do not corroborate the change in aurophilicity, but ultimately increase the ionization potential of the Au centre.9,10
Chemical modification, i.e., bond variation by ligand substitution or modification, is the established method for the manipulation, control and fine tuning of aurophilic Au(I) interactions in linear complexes of type [Au(L)2]+.11 However, chemical modification restricts the ability to manipulate solely the aurophilic interactions, due to the concomitant changes in other chemical bonds and groups around the Au(I) centres. The steric requirements of ligands affect the way in which the molecules pack, with bulkier groups reducing effective packing of molecules. In the absence of steric constraints crystal packing is determined primarily by the presence of the Au⋯Au contacts perpendicular to the gold–ligand axis: Au(I) complexes of primary phosphines form elongated chains of Au(I) centres, while secondary and tertiary phosphines generate binuclear species.12 A comparative study of the crystal structures of [AuI(PPh3)] and [AuI(PMe3)] revealed dimer formation in the latter through Au⋯Au contacts that are significantly shorter than in the former.13 Surprisingly, although high pressure crystallography offers a more versatile method of controlling and investigating aurophilic interactions by forcing Au(I) centres closer together, its potential remains largely untapped, as evidenced by the very small number of such reports in the literature. The single-component molecular metal [Au(tmdt)2] (tmdt = trimethylenetetrathiafulvalenedithiolate) reported by Kobayashi et al. in 2009 was the first crystallographic study of the properties of a gold complex as a function of pressure, although it features S⋯S rather than Au⋯Au contacts.14
The first systematic high pressure study into the relationship between aurophilicity and luminescent properties of Au(I) complexes appeared in 2014 with a series of four trimeric pyrazolate-based complexes.15 The observed red shifts of their luminescence on increasing pressure were correlated with changes in aurophilicity in these systems. In contrast, the lack of emission in complexes incorporating the sterically-demanding diphenylpyrazolato ligand was attributed to the absence of intermolecular aurophilic interactions under pressure, precluded by the bulk of the ligand. There is a general scarcity of high pressure studies of coordination complexes,16 not just of Au(I) species. As part of our focus on the chemistry of organogold complexes,17 we were interested in investigating the influence of pressure in modifying aurophilic interactions in [1,4-C6H4{PPh2(AuCl)}2] 1, a representative of a significant class of phosphine Au(I) halides which demonstrate significant luminescence.18 Properties such as emission are highly sensitive to the nature of the Au⋯Au interaction. Herein, we present the first high-pressure study for this family of Au(I) complexes, in which we employ pressure to investigate the nature of the Au⋯Au interaction in a controlled manner not possible using conventional synthetic chemical substitution approaches. As a complement to our crystallographic approach,19 Hirshfeld surface analysis, theoretical calculations and high pressure Raman spectroscopy of 1 were employed to advance our understanding of the effects of pressure on this model complex.
At ambient pressure, 1 crystallizes in the monoclinic space group C2/c with one half of the molecule defining the asymmetric unit and the central phenyl ring lying across an inversion centre (Fig. 1). The P–Au–Cl subunits have the expected linear geometry, P1–Au1–Cl1 179.11(8)°, and their disposition is close to mutually orthogonal. The P centre adopts a slightly distorted tetrahedral geometry with Au–P–Cl valence angles slightly larger than the ideal tetrahedral values. The Au1–P1 and Au1–Cl1 distances are 2.2256(16) Å and 2.2725(17) Å, respectively, and all these bond lengths and angles are comparable to previously reported values.20 At ambient pressure, the three-dimensional packing of the molecules of 1 involves long intermolecular interactions, including of types H⋯H, C–H⋯π, Au⋯Au and π⋯π, all of which play important roles in the behaviour of 1 under pressure. Adjacent molecules of 1 orientate themselves in a mutually trans arrangement, giving rise to the π⋯π interactions seen in the molecular packing (Fig. 2).
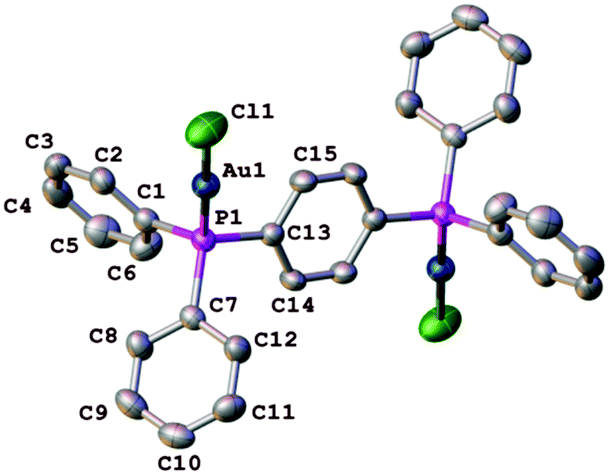 | ||
| Fig. 1 Structure of 1 comprising two asymmetric units related by a centre of inversion, at ambient pressure. H are atoms omitted for clarity and only the atoms of the asymmetric unit are labelled. Displacement ellipsoids are drawn at the 50% probability level. | ||
 | ||
| Fig. 2 Packing arrangement of 1 at ambient pressure showing (a) π⋯π interactions, which are highlighted by a blue circle and (b) aurophilic interactions, which are highlighted by a red ellipse. | ||
When 1 is placed under pressure there is, as expected, an overall compression of the unit cell parameters and volume with increasing pressure (Fig. S1, S2 and Table S1, ESI†). The unit cell volume contracts by 796.87(8) Å3, from 2769.37(8) Å3 at ambient pressure to 1972.5(2) Å3 at 106.2 kbar, an overall contraction of 29% and comparable to that seen in other high pressure studies of gold(I) complexes.15 The rate of compression decreases with increasing pressure, with the unit cell volume decreasing by 14% over the first 19.6 kbar, but only by 15% over the next 86.6 kbar, consistent with the remaining van der Waals space becoming much more difficult to compress. Fitting a third-order Birch–Murnaghan equation of state (Table S3, ESI†) gives a bulk modulus of 8(13) GPa for 1, comparable to other “soft” materials such as Ru3(CO)12.21
Anisotropy is clearly present in the rate of contraction of lattice parameters a, b and c, with overall compression being 9.3, 8.6 and 14.4%, respectively, over the pressure range studied. The smooth compression of all of these parameters (Fig. S1 and S2, ESI†) implies that there is no significant reorganization of the molecules as a function of pressure.
The structural changes with increasing pressure were investigated and selected bond lengths and angles are shown in the ESI† (Table S2). Over the pressure range studied, the bond distances and angles shift from their original values, resulting in the overall compression of the molecules to a denser, close-packed structure (Fig. S3, ESI†).
The presence of several types of intermolecular interactions is of particular interest in 1 and they are all affected significantly by pressure. The Au⋯Au interactions lie parallel to the c axis, which is consistent with the greater degree of compression observed in this direction. In contrast, the π⋯π interactions are not aligned parallel to any of the principal axes. At ambient pressure, the distance between Au centres in adjacent molecules is 3.6686(5) Å, corresponding to a relatively long Au⋯Au contact and longer than the sum of the van der Waals radii for two Au atoms (2.9 Å) (Fig. S4, ESI†).3 As pressure is increased, the aurophilic interaction shortens by 0.6132(13) Å, from 3.6686(5) Å at ambient pressure to 3.0554(12) Å at 106.2 kbar (Fig. S4, ESI†). The shortening of the Au⋯Au interaction is accompanied by the expected ligand bend-back, as observed in the deviation from linearity of the P1–Au1–Cl1 angle: the value of 179.11(8)° at ambient pressure falls to one of 172.62(13)° at 106.2 kbar.22 To the best of our knowledge, over the pressure range studied this is the largest pressure-induced contraction in the length of an aurophilic interaction in any Au(I) complex. CSD database searches (Fig. S5 and S6, ESI†) reveal that an Au⋯Au separation of 3.0554(12) Å lies at the lower end of the range of reported values for Au complexes studied at high pressure. Despite its shortness, there is no indication that an Au–Au chemical bond has actually formed: this would require the Au⋯Au distance to fall below 2.9 Å in order to lie within the sum of the covalent radii.3 The decreasing response of the Au⋯Au distance at the highest pressures suggests that this criterion would not be achieved for 1 even at substantially higher pressures.
π⋯π interactions also play an important role in the behaviour of 1 at high pressure. The centroid–centroid distance between adjacent phenyl rings related by a crystallographic two-fold axis is 3.728(3) Å at ambient pressure, decreasing by 0.695(7) Å to 3.031(6) Å at 106.2 kbar (Fig. S4, ESI†). The rate at which the π⋯π interaction contracts decreases with increasing pressure, falling by 8.2% from its original distance over the first 19.6 kbar, whilst a compression of only 11.5% is observed over the next 86.6 kbar. In contrast, the rate at which the Au⋯Au interaction contracts is more consistent (4.4% and 13%, respectively) over the same pressure ranges. 1 exhibits extensive ring overlap between adjacent phenyl rings even at ambient pressure, and this overlap increases with increasing pressure (Table S4 and Fig. S7, ESI†). The enhanced overlap of the adjacent phenyl rings and the significant shortening of the π⋯π interaction with pressure severely restrict further compression of the molecules beyond 106.2 kbar. At this pressure the phenyl rings are so close [centroid–centroid distance = 3.031(6) Å] that the π⋯π interactions become clearly repulsive in nature, thereby preventing the formation of shorter Au⋯Au interactions.
Excluding the short Au⋯Au distance we have identified, there are no close Au⋯Au contacts below ca. 8 Å at ambient pressure or below ca. 6.5 Å at 106.2 kbar.
Hirshfeld surface analysis23 of the surfaces and contacts for 1 reveals the presence of several types of intermolecular interaction; the number of different contacts increases with pressure (Tables S5 and S6, ESI†). At ambient pressure, there are few close contacts (represented by the red areas in Fig. 3a). The most pronounced red area can be assigned as a π⋯π interaction with an adjacent molecule. As expected, with increasing pressure more close contacts (red areas) appear on the surface as the molecules are forced closer together. The additional red areas can be assigned to whole range of different intermolecular interactions but the most prominent areas relate to π⋯π, C–H⋯π, π⋯C–H and H⋯H interactions as highlighted in Fig. 3b. These prominent red areas show excellent correspondence with the shortening of the intermolecular interactions; it is not just the Au⋯Au and π⋯π interactions that shorten considerably.
 | ||
| Fig. 3 Hirshfeld surface of 1 at (a) ambient pressure and (b) 106.2 kbar. Red areas: contacts which are shorter than the sum of the van der Waals radii; white areas: contacts which are short but non-overlapping; blue areas: contacts which are longer than the sum of the van der Waals radii. The coloured circles represent the different types of interaction: π⋯π (black); C–H⋯π (green); π⋯C–H (red); H⋯H (blue). | ||
The fingerprint plots24 at ambient pressure and 106.2 kbar show a large surface area compared with previously reported examples,25 and these span a large range of distances from 1.2 to 2.6 Å at ambient pressure and 0.8 to 2.2 Å at 106.2 kbar (Fig. 4), suggesting that several types of intermolecular interaction are present. It is noteworthy that the shape and position of these plots also changes with increasing pressure. By 106.2 kbar, the position of the whole plot has shifted considerably to shorter distances, confirming that a denser, more close-packed structure is formed at higher pressures. The shapes of the plots at 106.2 kbar and at ambient pressure are clearly different (see Fig. S9–S13, ESI†). At ambient pressure, there are two spikes at the top left and bottom right of the plot, which correspond to the shortest C–H⋯π distance of 2.845(6) Å at ambient pressure. Notably, these spikes are not present in the fingerprint plot at 106.2 kbar, suggesting that other interactions now dominate the crystal packing. The spike along the diagonal at 106.2 kbar suggests the presence of close head-to-head H⋯H contacts. Supporting evidence is available in the crystallographic data, where H⋯H interactions occupy the same plane (Fig. S8, ESI†). The length of this H⋯H interaction decreases from 2.54 Å at ambient pressure to 1.94 Å at 106.2 kbar.
 | ||
| Fig. 4 Fingerprint plots of a molecule of 1 at (a) ambient pressure and (b) 106.2 kbar, showing all the intermolecular interactions present. The red circle highlights closer Au⋯Au interactions. | ||
DFT calculations were carried out on a model of 1 in order to provide insight into the energetics associated with the intermolecular interactions between two molecules of 1 as a function of pressure. At ambient pressure, the bonding energy ΔEbond is −22.91 kJ mol−1, indicative of an attractive interaction. Beyond 19.6 kbar, ΔEbond becomes positive and increases to 86.0 kJ mol−1 at 106.2 kbar (Table S7, ESI†), confirming that repulsive energies contribute more to ΔEbond: this result is consistent with the increasing difficulty of compressing the van der Waals space at higher pressures.
In order to further characterize the response of 1 to pressure, Raman spectroscopy was carried out in a DAC. Jones et al. reported26 the vibrational frequencies of triphenylphosphine gold(I) halides and assigned the bands at 329 and 182 cm−1 to ν(Au–Cl) and ν(Au–P) stretching modes, respectively. Raman investigations of 1 showed a characteristic vibration at 160 cm−1 which shifts linearly to higher energy with increasing pressure (Fig. S18, ESI†): this can be tentatively assigned as a ν(Au–P) stretching vibration (Fig. S19, ESI†). There is also a strong band at 330 cm−1 which can be assigned as ν(Au–35Cl) (Fig. S20, ESI†), while the shoulder at 323 cm−1 is characteristic of ν(Au–37Cl). Again, this band shifts linearly to higher energy with increasing pressure, consistent with the observed compression of the bond length at similar pressures (Fig. S18, ESI†). Aromatic ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) stretching frequencies can be assigned to the features at 1587 cm−1 which similarly shift slightly to higher energy with increasing pressure (Fig. S21, ESI†).27 Perreault et al. found evidence for the presence of aurophilic interactions in Au2 dimers from Raman spectroscopy:28 their extensive study suggests that ν(Au2) frequencies lie between 30 and 200 cm−1 depending on the ligand substituents, the metal–metal distance and the metal–metal force constants. In the Raman spectrum of 1 (Fig. 5) there is a strong peak at 110 cm−1 that shifts to higher energy on increasing the pressure, which we tentatively assign as ν(Au2). This peak is in good agreement with literature values29 and behaves in line with Perreault's observations of increasing ν(Au2) frequency with decreasing Au⋯Au distance across a range of gold dimer complexes. More notably, using Perreault's calculations and our peak positions to predict the force constants and hence the Au⋯Au distance, we obtain a value of 2.783 Å at ambient pressure and 2.566 Å at 81.2 kbar. These values are rather shorter than those we observe crystallographically, which supports our commentary on the intermolecular interactions inhibiting the shorter contact between the metal centres.
C) stretching frequencies can be assigned to the features at 1587 cm−1 which similarly shift slightly to higher energy with increasing pressure (Fig. S21, ESI†).27 Perreault et al. found evidence for the presence of aurophilic interactions in Au2 dimers from Raman spectroscopy:28 their extensive study suggests that ν(Au2) frequencies lie between 30 and 200 cm−1 depending on the ligand substituents, the metal–metal distance and the metal–metal force constants. In the Raman spectrum of 1 (Fig. 5) there is a strong peak at 110 cm−1 that shifts to higher energy on increasing the pressure, which we tentatively assign as ν(Au2). This peak is in good agreement with literature values29 and behaves in line with Perreault's observations of increasing ν(Au2) frequency with decreasing Au⋯Au distance across a range of gold dimer complexes. More notably, using Perreault's calculations and our peak positions to predict the force constants and hence the Au⋯Au distance, we obtain a value of 2.783 Å at ambient pressure and 2.566 Å at 81.2 kbar. These values are rather shorter than those we observe crystallographically, which supports our commentary on the intermolecular interactions inhibiting the shorter contact between the metal centres.
 | ||
| Fig. 5 Variation in the ν(Au2) stretching frequency (identified by an asterisk) with increasing pressure. | ||
We have shown that high pressure crystallography offers a means to manipulate and modify the aurophilic interactions in Au(I) complexes, beyond what is feasible by chemical substitution. We can thereby investigate the Au⋯Au interaction in a controlled manner. We have also confirmed that the application of pressure can have major effects on these aurophilic interactions. A combination of structural control via high pressure crystallography and structural design by chemical modification offers a potential future route to greater compression of the Au⋯Au distance, allowing the controlled formation of Au–Au bonds that can be characterised both structurally and spectroscopically.
NM is grateful to the Australian Government for receipt of an Endeavour Research Fellowship. AEOC acknowledges support from the University of Nottingham for a PhD studentship and thanks the Nottingham Nanotechnology and Nanoscience Centre for access to Raman spectroscopy facilities. TLE thanks the Royal Society for the award of a University Research Fellowship. We thank Dr Steven H. Privér for the synthesis of 1,4-[C6H4{PPh2(AuCl)}2] and Dr Jonathan McMaster for helpful discussions. We are grateful to EPSRC (award EP/K038869/1) and the University of Nottingham for support. MS acknowledges receipt of an ERC Advanced Grant. We thank Dr Ahmad Kandjani for designing the graphical abstract.
Notes and references
- H. Schmidbaur, Chem. Soc. Rev., 1995, 24, 391 RSC; C. Latouche, Y.-R. Lin, Y. Tobon, E. Furet, J.-Y. Saillard, C.-W. Liu and A. Boucekkine, Phys. Chem. Chem. Phys., 2014, 16, 25840 RSC.
- E. R. T. Tiekink, Coord. Chem. Rev., 2014, 275, 130 CrossRef CAS.
- H. Schmidbaur, Gold Bull., 1999, 33, 3 CrossRef.
- P. G. Jones, Gold Bull., 1981, 14, 102 CrossRef CAS.
- P. Pyykkö and Y. Zhao, Angew. Chem., Int. Ed., 1991, 30, 604 CrossRef.
- P. Pyykkö, Angew. Chem., Int. Ed. Engl., 2004, 43, 4412 CrossRef PubMed.
- M. Barysz and P. Pyykkö, Chem. Phys. Lett., 2000, 325, 225 CrossRef CAS.
- N. Runeberg, M. Schütz and H.-J. Werner, J. Chem. Phys., 1999, 110, 7210 CrossRef CAS.
- E. O'Grady and N. Kaltsoyannis, Phys. Chem. Chem. Phys., 2004, 6, 680 RSC.
- A. Otero-de-la-Roza and J. D. Mallory, J. Chem. Phys., 2014, 140, 18A504 CrossRef PubMed.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2008, 37, 1931 RSC; H. Schmidbaur, A. Grohmann and M. E. Olmos, in Gold: Progress in Chemistry, Biochemistry and Technology, ed. H. Schmidbaur, John Wiley & Sons, Chichester, 1999 Search PubMed; F. Mohr, Gold Chemistry, Applications and Future Directions in the Life Sciences, John Wiley & Sons, 2009 Search PubMed.
- H. Schmidbaur, Gold Bull., 1990, 23, 11 CrossRef CAS; H. Schmidbaur, G. Weidenhiller, O. McAuliffe, R. G. Pritchard, R. Fields and B. Beagley, J. Chem. Soc., Dalton Trans., 1989, 907 Search PubMed.
- S. Ahrland, K. Dreisch, B. Norén and Å. Oskarsson, Acta Chem. Scand., Ser. A, 1987, 41, 173 CrossRef.
- Y. Okano, B. Zhou, H. Tanaka, T. Adachi, Y. Ohishi, M. Takata, S. Aoyagi, E. Nishibori, M. Sakata, A. Kobayashi and H. Kobayashi, J. Am. Chem. Soc., 2009, 131, 7169 CrossRef CAS PubMed.
- C. H. Woodall, S. Fuertes, C. M. Beavers, L. E. Hatcher, A. Parlett, H. J. Shepherd, J. Christensen, S. J. Teat, M. Intissar, A. Rodrigue-Witchel, Y. Suffren, C. Reber, C. H. Hendon, D. Tiana, A. Walsh and P. R. Raithby, Chem. – Eur. J., 2014, 20, 16933 CrossRef CAS PubMed.
- J. P. Tidey, H. L. S. Wong, M. Schröder and A. J. Blake, Coord. Chem. Rev., 2014, 277, 187 CrossRef.
- N. Mirzadeh, M. A. Bennett and S. K. Bhargava, Coord. Chem. Rev., 2013, 257, 2250 CrossRef CAS.
- E. R. T. Tiekink and J.-G. Kang, Coord. Chem. Rev., 2009, 253, 1627 CrossRef CAS.
- Crystallographic information for 1 at different pressures can be found in Table S1 (ESI†) and in CCDC 1429742–1429755.
- S. P. C. Dunstan, P. C. Healy, A. N. Sobolev, E. R. T. Tiekink, A. H. White and M. L. Williams, J. Mol. Struct., 2014, 1072, 253 CrossRef CAS.
- C. Slebodnick, J. Zhao, R. Angel, B. E. Hanson, Y. Song, Z. Liu and R. J. Hemley, Inorg. Chem., 2004, 43, 5245 CrossRef CAS PubMed.
- A. Ilie, C. I. Raţ, S. Scheutzow, C. Kiske, K. Lux, T. M. Klapötke, C. Silvestru and K. Karaghiosoff, Inorg. Chem., 2011, 50, 2675 CrossRef CAS PubMed.
- J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 627 CrossRef PubMed.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19 RSC.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378 RSC.
- A. G. Jones and D. B. Powell, Spectrochim. Acta, Part A, 1974, 30, 563 CrossRef.
- M. Kumar, M. Srivastava and R. A. Yadav, Spectrochim. Acta, Part A, 2013, 111, 242 CrossRef CAS PubMed.
- D. Perreault, M. Drouin, A. Michel, V. M. Miskowski, W. P. Schaefer and P. D. Harvey, Inorg. Chem., 1992, 31, 695 CrossRef CAS.
- R. J. H. Clark, J. H. Tocher, J. P. Fackler Jr., R. Neira, H. H. Murray and H. Knackel, J. Organomet. Chem., 1986, 303, 437 CrossRef CAS; T. F. Carlson and J. P. Fackler Jr., J. Organomet. Chem., 2000, 596, 237 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1429742–1429755. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc00923a |
| This journal is © The Royal Society of Chemistry 2016 |