
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploiting redox activity in metal–organic frameworks: concepts, trends and perspectives
D. M.
D'Alessandro
School of Chemistry, The University of Sydney, New South Wales, 2006, Australia. E-mail: deanna.dalessandro@sydney.edu.au
First published on 8th March 2016
Abstract
Of the many thousands of new metal–organic frameworks (MOFs) that are now discovered each year, many possess potential redox activity arising from the constituent metal ions and/or organic ligands, or the guest molecules located within their porous structures. Those redox states that can be accessed via postsynthetic redox modulation often possess distinct physical properties; if harnessed, these provide a basis for applications including microporous conductors, electrocatalysts, energy storage devices and electrochemical sensors, amongst others. This feature article highlights the latest developments in experimental, theoretical and computational concepts relevant to redox-active MOFs, including new solid state electrochemical and spectroelectrochemical techniques that have great utility in this field. A particular emphasis is on current and emerging trends at the fundamental level which underscore the importance of this promising class of electroactive materials for a wide range of technologically- and industrially-relevant applications.
1. Introduction
The development of redox-active MOFs is a highly sought after goal: at a fundamental level these materials offer unprecedented insights into charge transfer in three-dimensional coordination space; at an applied level, their properties may underpin the next generation of technologically useful devices. Over the past 20 years, MOFs1,2 have developed at an extraordinary pace owing to the enormous structural and chemical diversity of these highly porous crystalline materials which has spurred myriad potential applications in gas storage, separation and catalysis, amongst others.3 The potential to exploit the redox-active properties of MOFs is an aspect that has gained attention relatively recently. This can be attributed to the fact that the metal centres (e.g., d10 ZnII) and organic linkers (e.g., carboxylates) traditionally used for MOF construction are often redox-inactive.The redox chemistry of molecular transition metal complexes is an area of significant fundamental interest and importance (e.g., the use of model systems for understanding charge transfer phenomena in nature). For redox-active MOFs, new opportunities are present to explore, for the first time, deeply fundamental aspects of charge transfer within three-dimensional coordination space. In contrast to traditional classes of polymeric and solid state materials such as alloys, metal oxides, mesoporous carbons and zeolites, the molecular nature of MOFs enables an exquisite level of control over their structural, chemical and physical properties at the nanoscale. The presence of cavities and windows due to the crystalline array of nanopores also provides an unprecedented opportunity to exploit host–guest chemistry. The properties of MOFs in their different redox states may be expected to vary significantly due to distinct structural, electronic, magnetic, fluorescent and host–guest properties, amongst others.4
The earliest investigations on a redox-active framework material (although not strictly a MOF) are arguably those on Prussian blue, [FeIII4{FeII(CN)6}3]·xH2O, which has been prized industrially for centuries as an ink and dye-stuff. Its “redox isomers” Berlin green (the all-FeIII analogue) and Prussian white (all-FeII version) can be prepared de novo or generated from Prussian blue itself by electrochemical or spectroelectrochemical methods.5 Despite some ambiguities regarding the exact composition of FeII and FeIII in various preparations of Prussian blue, the ability to switch between the different redox states has been exploited to modulate the conducting, magnetic, gas adsorption, and optical properties.5–8
The historically rich field of metal cyanide frameworks such as Prussian blue offer key insights into the fundamental importance of redox activity in MOFs, and the enormous value of harnessing this property for functional applications. Some important lessons relevant to the broader field of redox-active MOFs are also apparent. These include the need for redox states that are accessible via chemical or electrochemical modulation, and the need for stability of these states on the timescale required for measurement. Generating bulk quantities of MOFs in their distinct electronic states requires appropriate chemical oxidants or reductants that do not cause degradation. Altering the redox state of a framework may have additional structural consequences: (i) it drives the incorporation of a counter-ion for charge balance, and (ii) it may instigate a structural change in the metal- or ligand-based components. In the first case, counter-ion inclusion/expulsion impacts the pore space, and the size and charge of this guest species must be considered in relation to the window aperture and pore size of the framework. Furthermore, despite the addition of a stoichiometric quantity of a chemical oxidant/reductant, the actual extent of the redox change in the framework itself may be limited to surface confined processes, and characterisation must be carefully undertaken. In the second case, MOFs constructed from first-row transition metal centres are notoriously prone to changes in geometry upon redox-state changes, and for this reason, addressing redox activity in such materials may not be possible. In the case of redox-active ligands, radical states (particularly anion radicals) are often sensitive to atmospheric oxygen, making the use of an inert atmosphere for synthesis necessary.
For applications which require a sufficient quantity of MOF, bulk synthesis of the material in a given redox state is required. Meanwhile, in situ methods for generating redox states via electrochemical means are valuable at the fundamental level for initial measurement of potential redox-accessible states. Recently, in situ spectroelectrochemical methods have proven their utility for understanding the fundamental spectroscopic properties of the distinct redox sates.9
This article focuses on key experimental, theoretical and computational concepts relevant to redox-active MOFs and highlights current trends in their study at both the fundamental and applied levels. A particular emphasis is how redox activity can be exploited to elucidate charge transfer phenomena, and at the applied level, how the redox-accessible states can be exploited to engender functional properties of technological and industrial importance. Many materials, including ionic MOFs10,11 for example, may have the potential to exhibit redox activity, but this may not have been probed. The scope is therefore restricted to cases where studies have investigated more than one redox state. Attention is also focused on intrinsic redox activity involving electron or hole movement through the framework backbone,12 rather than ionic conduction through the pores.13–15 The discussion here is also restricted to redox transformations that retain the structure of a framework, however, it is interesting to note that amorphous solids derived from “MOF precursors” do exhibit highly desirable functional properties.13 For ease of readability, a list of acronyms for ligands is provided at the end of this article.
2. Fundamental concepts
The highly ordered crystalline structures of MOFs coupled with the potential for systematic tuning of their structural properties offers a unique platform for probing deeply fundamental aspects of charge transfer in multi-dimensional coordination space. Various synthetic methods have been employed to generate redox-active MOFs as single crystals and bulk solids including traditional solvothermal methods and electrosynthesis,16,17 amongst others.18,19 Recent developments in fabrication methods for producing highly ordered and oriented thin films20–24 on surfaces such as conductive substrates has further accelerated the pathway for integrating redox-active MOFs into device architectures.25–272.1 Engendering redox activity in MOFs
Redox-active MOFs fall in two general classes, where (i) redox-active metals or ligands are present in the as-synthesised framework, or (ii) the redox-active molecules (generally organic species) are introduced postsynthetically into redox-inactive frameworks by covalent bonding or impregnation as guests.18 In the first case, the origin of the activity arises either from metal centres (which do not alter their geometries upon redox state changes) or ligands (which often possess stable radical states). Although outside the scope of this discussion, even in cases where framework degradation occurs following a redox change, this structural instability has been exploited to produce amorphous materials which have shown promise as electrocatalysts.28 In cases where the metal centres are magnetically interesting or the ligands possess potential radical states, redox modulation offers a pathway for these materials to be employed as spin probes and in magnetic switching devices.29 While redox state changes may necessitate the concomitant introduction or expulsion of counterbalancing charges (typically in the form of counter-anions or cations), the remaining pore space can be exploited for functional applications, as discussed in Section 3.In the second class of redox-active MOFs, the postsynthetic infiltration of redox-active species into redox-inactive frameworks takes advantage of the high affinity of MOFs for non-polar and polar guest molecules. In some cases, redox modulation of the guest induces a change in the electronic properties of the framework, as illustrated by the incorporation of redox-active metallocenes into [VO(bdc)] which induced partial framework reduction.30 An attractive application for this class of materials is in redox driven release mechanisms.18 For example, [Zn4O(bdc)(btb)4/3], otherwise known as UMCM-1, containing alizarin red S or methylene blue as guests exhibited potential driven uptake and release of charge compensating protons upon switching the redox state of the guest.31,32 Postsynthetic ion metathesis techniques have also proven successful for the introduction of redox-active first-row transition metal ions including TiIII, VII/III, CrII/III, MnII and FeII into the redox-inactive MOF-5, [Zn4O(bdc)], scaffold.33 The synthesis of these metallated analogues could not be achieved using a de novo route. This work provided the first evidence for the redox activity of MOF-5 analogues, with the achievement of the stoichiometric single oxidation of CrII-MOF-5, and the discovery that FeII-MOF-5 activates NO via electron transfer from the Fe centre.34
The covalent grafting of redox-active species such as metallocenes into redox-inactive frameworks has been shown to improve the stability of host–guest systems during redox cycling. A number of frameworks have been targeted in this regard including [Zn(Fcdc)(bpy)],35 [Zn4O(bdc-NH2)(btb)4/3] (UMCM-1-NH2),36 [Al(OH)(bdc)] (MIL-47(Al)),37 and the NU-1000 framework consisting of [Zr6(μ3-O)4(μ3-OH)4(OH)4(OH2)4]8+ secondary building units linked by 1,3,6,8-tetrakis(p-benzoic acid)pyrene (H4TBAPy) ligands.38 Redox hopping between the anchored redox-active groups (e.g., ferrocenyl/ferrocene, Fc+/Fc) was suggested as the predominant mechanism for charge transport. A particularly interesting observation was the dependence of the permselectivity of ferrocene-functionalised NU-1000 thin-films on the electrolyte concentration: films were permeable to solution cations when the MOF was in the ferrocenium form, but largely impermeable in the ferrocene form.38 The mechanisms for charge transport within MOF films have been suggested to involve linker9,39–41 or shuttle-to-shuttle38 electron/hole redox hopping.42 The importance of counter-ion diffusion was noted for [Zn(Fcdc)(bpy)],35 where the redox processes were found to be controlled by the movement of charge compensating counter-anions into the framework upon oxidation of the ferrocene units.
2.2 Redox activity and charge transfer MOFs
A fundamental issue for redox-active MOFs is the question of how charge propagates through the crystal lattice. The mechanisms can be broadly classed as through-bond or through-space and may involve hole or electron transfer. In cases where porosities are low, charge transport is likely to involve surface-confined hopping processes39 where the redox centres on the surface of MOF particles undergo redox changes while the bulk remains unaffected. This hopping behaviour is also expected for insulating frameworks where the electroactive components are well separated spatially and where effective overlap of frontier orbitals does not exist.Effective overlap of frontier orbitals and an optimal spatial arrangement of components facilitates charge delocalisation, leading to a conductive framework. An important consideration here is the introduction of counter-ions needed to balance framework charge, which necessarily impedes the porosity.
In addition to the aforementioned strategies for the design of redox-active MOFs, some specific modes of charge transport are also central to the discussion (see Fig. 1).27,43–45 These modes can involve through-bond charge transfer in the case of highly delocalised systems or through-space transfer by virtue of the close proximity of components. Redox-active moieties are important for facilitating long-range electron transfer, as is already known for one and two-dimensional conductors,12 however it is important to note that charge delocalisation can take advantage of the organic ligands themselves, with redox-inactive metal centres merely providing “structural support”. This π-stacking mediated transfer is exemplified by [Zn2(TTFTB)(H2O)2] (Fig. 1a) incorporating benzoate functionalised tetrathiafulvalene (TTF) ligands. Charge transfer occurs along infinite TTF stacks resulting in high electron mobility.46,47 Indeed, one of the earliest known examples of a coordination polymer exhibiting metallic-like conductivity was [Cu(Me2-DCNQI)2] which featured ligand π–π stacks.48
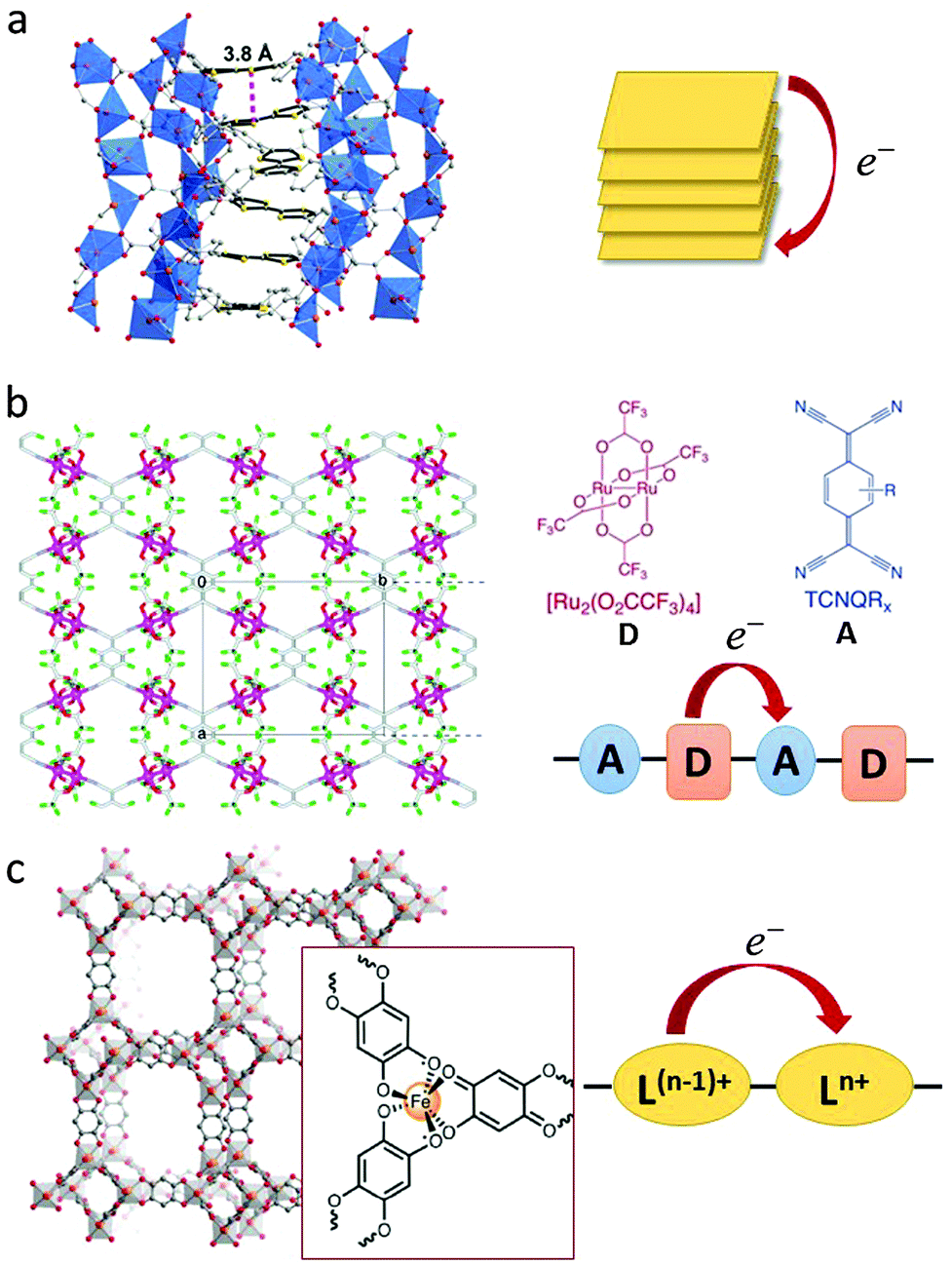 | ||
| Fig. 1 Examples of strategies for through-bond or through-space charge transfer interactions in MOFs. (a) Side view of a helical TTF stack with a depiction of the shortest intermolecular S–S contact in [Zn2(TTFTB)(H2O)2]. Adapted with permission from ref. 46. Copyright 2012 American Chemical Society. (b) Through-bond donor–acceptor charge transfer in [{Ru2(F4PhCO)4}2(TCNQ)]. Adapted with permission from ref. 58. Copyright 2010 American Chemical Society. (c) Through-bond mixed valency leading to Intervalence Charge Transfer in [(NBu4)2FeIII2(dhbq)3]. The tris-chelating iron centre is highlighted in the insert. Ln+ and L(N−1)+ represent the ligand in its distinct redox states. Adapted with permission from ref. 63. Copyright 2015 American Chemical Society. | ||
In two-dimensional graphene-like MOFs such as [Ni3(hitp)2]49 and [Ni3(bht)2]50 strong π-conjugation supports electron delocalisation through the two-dimensional sheets. Here, there is significant overlap of the metal and ligand frontier orbitals, thus facilitating conductivity.
Two other modes of charge transport can be broadly termed donor–acceptor mechanisms, the origins of which have long and rich historical foundations. In the first case, organic charge transfer (CT) salts constructed purely from organic components have been the source of widespread experimental and theoretical interest for over four decades owing to the exquisite tunability of their electronic structures, and their numerous potential applications in devices such as field effect transistors, photovoltaic cells and light emitting diodes, amongst others.51–53 The most conductive of these are the so-called “organic metals” including archetypal TTF-TCNQ.54 Key to CT phenomena in these systems is the presence of components that can possess stable (neutral, anionic or cationic) radical states. Electronic coupling between the highest occupied molecular orbital (HOMO) of the donor (D) and the lowest unoccupied molecular orbital (LUMO) of the acceptor (A) generates a partial degree of CT (δ) between D and A (also known as the Madelung energy, M), leading to a ground state that is characterised by a partial ionicity.51,53 The magnitude of δ is dictated by the relative ionisation potential of D (ID) and electron affinity of A (EA), where M(δ) ≈ ID − EA. Three possibilities may arise depending on the magnitude of δ: a neutral or quasi-neutral state (δ < 0.5), a partial CT or mixed-valence state (0.5 ≤ δ < 1) state, or a fully ionic state (δ = 1). A plot of the energy of the CT band maximum (ΔECT) versus the potential energy difference between D and A results in the famous Torrance diagram which exhibits a clear boundary between the neutral and ionic states.55,56 Furthermore, the occurrence of a partial CT state (known as the ‘mixed-valence regime’) is predicted to generate conductive or metallic-like coordination solids.57 In addition to these electronic contributions, the structural characteristics of the components (e.g., their degree of planarity) and the arrangement of D and A will also have a critical influence on δ. The degree of CT controls numerous physical properties of CT salts including the lattice energies, bond lengths, optical band gaps and conductivities. Empirical relationships between intrinsic properties of the organic components have also been described using ionicity phase diagrams for the most common CT complexes.51
In contrast to organic CT, the understanding of charge transfer interactions in redox-active MOFs is in its infancy. In addition to their increased dimensionality and structural robustness relative to the organic systems, the potential void space in MOFs is highly advantageous for explorations of guest-induced interactions. The first evidence for the applicability of the semiempirical Torrance diagram to coordination solids was reported for through-bond CT in a series of two- and three-dimensional frameworks incorporating carboxylate-bridged paddlewheel-type diruthenium(II,II) units, [RuII,II2(F4PhCO)4] bridged by TCNQ˙− and DCNQI˙− radical anions.58 In these systems, a systematic dependence of the charge transfer energy on the HOMO–LUMO gap of the D (Ru2) and A (ligand) was reported.59
Two recent examples of CT frameworks demonstrate the potential to exploit through-bond or through-space CT mechanisms. In the first case, infiltration of the electron acceptor TCNQ into [Cu3(btc)2] (HKUST-1), led to a bridging of the dinuclear copper paddlewheel clusters with the ligands and their partial reduction.45,60,61 The result was strong through-bond electronic coupling between the units and a concomitant increase in conductivity.61 By contrast, in the system [(Zn(DMF))2(TTFTC)(DPNI)], through-space CT interactions have been observed between the electron donor TTF and acceptor DPNI ligands.62
A second type of donor–acceptor interaction occurs for a partial CT state (0.5 ≤ δ < 1) termed a mixed-valence state, where D and A are the same moiety (either metal- or ligand-based) and are present in different formal oxidation states. A number of three-dimensional frameworks which exhibit mixed-valence states based on the metal centres have been reported.64 Examples include the pillared bilayer framework consisting of a bismacrocyclic NiII complex bridged by 1,3,5-benzetricarboxylate ligands, in which intercalation of iodine generates a mixed-valence material containing NiII/NiIII and I2/I3.65 Certain frameworks have also been prepared directly in mixed-valence form, as exemplified by transition metal cyanide compounds.
For archetypal cyanide-based materials such as Prussian blue, the importance of the mixed-valence phenomenon was first elucidated in the early twentieth century with the realisation that the odd electron was in rapid oscillation between the redox-active FeIII and FeII centres (rather than being uniquely fixed, one to each ion), giving rise to the absorption of light in the visible or near-infrared (NIR) region of the electromagnetic spectrum.66 According to the Robin and Day classification scheme,8 Prussian blue is an example of a “localised” Class II system in which the metal ions are weakly-coupled electronically leading to electron hopping.67 Two additional classes of mixed-valence systems are distinguished on the basis of the extent of charge transfer: Class I systems are characterised by non-interacting centres, and “delocalised” Class III systems by strongly-coupled centres. Electronic delocalisation is promoted by mixing between the donor and acceptor wave functions, and in the limit of strong overlap, delocalisation is complete and the metal centres possess identical valences (i.e., the system is conducting).
To date, mixed-valence mechanisms in MOFs have been suggested in a very limited number of cases. A rare example of organic mixed valency was recently reported in the framework [(NBu4)2FeIII2(dhbq)3] (Fig. 1c).63 Here, the ligands are present in both their quinone and semiquinone forms (dhbq2−/3−), giving rise to an Intervalence Charge Transfer (IVCT) transition. Taking inspiration from the rich literature on mixed valency in dinuclear complexes, conditions for the generation of metal-based mixed-valence MOFs are feasible,44 as is the possibility of generating mixed valency in MOFs via through-space intra-ligand IVCT of the type observed previously in molecular rectangles.68
2.3 Spectroscopic methods as a window to redox activity
An important consequence of CT interactions in MOFs is the occurrence of new features in their electronic absorption spectra that are distinct from those of the isolated components themselves. CT interactions of either the through-bond donor–acceptor type,61 the mixed-valence type,63 or the through-space donor–acceptor type,62 are manifested by CT bands in the visible and NIR regions of the electronic absorption spectra. Critically, their analysis provides fundamental information regarding the nature of CT.Compared with the rare example of the [(NBu4)2FeIII2(dhbq)3] framework in which an IVCT band was observed,63 CT in organic mixed-valence systems69 and dinuclear mixed-valence complexes based on second-row transition metal centres has been studied comprehensively.66,70–74 The mixed-valence Prussian blue framework has also been the subject of extensive investigations, and the rate of electron transfer has been calculated from its IVCT band by extracting the electronic coupling and reorganisational energy through application of the Marcus–Hush equation. Some evidence for systematic changes in the IVCT band have also been observed for the ruthenium analogue K1.2[Ru3.6{Ru(CN)6}3] which exhibited a lower energy band and an increased conductivity, presumably owing to the presence of the second-row transition metals with more diffuse d-orbitals.75 These spectroscopic signatures of CT may thus provide a window to functional properties of MOFs such as their conductivity. Despite the lack of analyses of CT in MOFs, enormous potential clearly exists to systematically probe these interactions by generating isostructural series of frameworks where the metal centres or organic ligands can be modulated to elucidate structure–function relationships.
2.4 Probing redox activity in MOFs
Solid state electrochemistry (including cyclic and square wave voltammetry) offers the primary tool for interrogating potential redox-accessible states of MOFs, and can provide details on the kinetics and mechanism of charge transfer reactions.18 Compared with solution state measurements, the electrochemistry of MOFs poses additional challenges due to the more complex processes associated with the various electrode-material and material–electrolyte interfaces, and the intricate ion diffusion processes which are coupled to charge transfer.76 For example, strong evidence has been found for the dependence of the electrochemical signal of the CuII/I couple in HKUST-1 on the size of the electrolyte anions/cations (i.e., the current for the CuII/I couple increased as the size of the cation decreased).77While many reported electrochemical measurements on solid materials have used composites with carbon paste, its use as a conductive supporting matrix precludes accurate analyses of electron transfer parameters. By contrast, relatively simple powder abrasion techniques have proven versatile for adhering solid materials to electrode surfaces and have enabled redox mechanisms to be derived,78,79 particularly cases that involve charge hopping between redox centres.35,39
Where MOFs are insulating and porosities are low, as noted for the ferrocenyl-modified MOF [Zn4O(bdc-NH2)1−x(bdc-NHC(O)Fc)x(btb)4/3], surface-confined electron hoping behaviour is expected.32 Since bulk ion diffusion through the framework was impeded, the peak currents were low relative to the amount of sample, and electron transport was fast leading to a scan rate independence of the peak potentials.
Recently, the application of slow scan cyclic voltammetry (using scan rate of 30 μV s−1 compared with rates typically above 5 mV s−1) was found to be particularly useful for the reductive insertion of Li+ (from a LiBF4 electrolyte) into the framework [(NBu4)2FeIII2(dhbq)3].63 This method has great future utility for addressing MOFs more broadly, including those assumed to be completely insulating.
Methods to achieve ex situ oxidation or reduction involve the use of chemical reagents such as halogens (e.g., Cl2, Br2 and I2), alkali metal salts (e.g., Li or Na napthalenide) or nitrosonium salts amongst others. Matching an appropriate chemical agent to a given redox transformation in a solid is non-trivial, often requiring screening to optimise the conditions. A recent elegant example of the non-innocent nature of redox-active ligands was reported for the framework [Mn2(dobdc)] (MOF-74) in which addition of a chemical oxidant led to selective oxidation of the dobdc4− ligand to the quinine form dobdc2−, whilst maintaining the MnII oxidation state of the metal centre.80
To address some of the issues associated with chemical methods for oxidation/reduction, in situ solid state spectroelectrochemical techniques based on UV-vis-NIR, EPR and Raman spectroscopies have recently been developed and successfully applied to MOFs.9,41,62 In these cases, the spectral response of the material is monitored in real time as a function of the applied potential, with the latter gauged from preliminary electrochemical measurements. Compared with chemical methods for oxidation/reduction, these techniques allow for the relatively rapid identification of the spectral properties of all redox-accessible states of a material, aiding the assignment of the origins of redox processes in electrochemistry based on their spectral signatures. Short lived, or relatively unstable redox-accessible states can thus be identified (e.g., those involving radical anions which are often unstable in the presence of an aerobic environment). Moreover, observation of stable isosbestic points and reversibility of the redox transformations support the retention of structure throughout the process. An important avenue of future work is in situ structural determination techniques which provide information on material stability throughout redox transformations.
In the same way that solution state in situ spectroelectrochemical methods have proven integral to interrogating fundamental charge transfer properties of soluble molecular species,81,82in situ solid state UV/vis/NIR, EPR and Raman spectroscopies have now proven their utility for MOFs, particularly in cases where materials degraded during attempts to perform chemical reduction/oxidation.62,83 Solid state UV/vis/NIR spectroelectrochemistry was first applied to the redox-active framework [Zn2(ndc)2(DPNI)], where the spectral signatures of the radical anion and dianion states of the DPNI ligand were observed as a function of the applied potential (Fig. 2).9 A range of other MOFs have also been interrogated using spectroelectrochemical methods, particularly those incorporating the TTF functionality, an organosulfur moiety that undergoes two reversible one electron oxidations localised on separate rings,84,85 and NDI, a well-studied electron acceptor.86 In the case of TTF, a partially oxidised ring has been shown to vastly improve conductivity,87 while functionalised versions of NDI often display unique electronic, electrochemical and optical properties.88 The utility of spectroelectrochemical techniques for addressing mechanisms of CT and understanding charge transport mechanisms in thin films of MOFs relevant to electrocatalysis has also recently been demonstrated.39
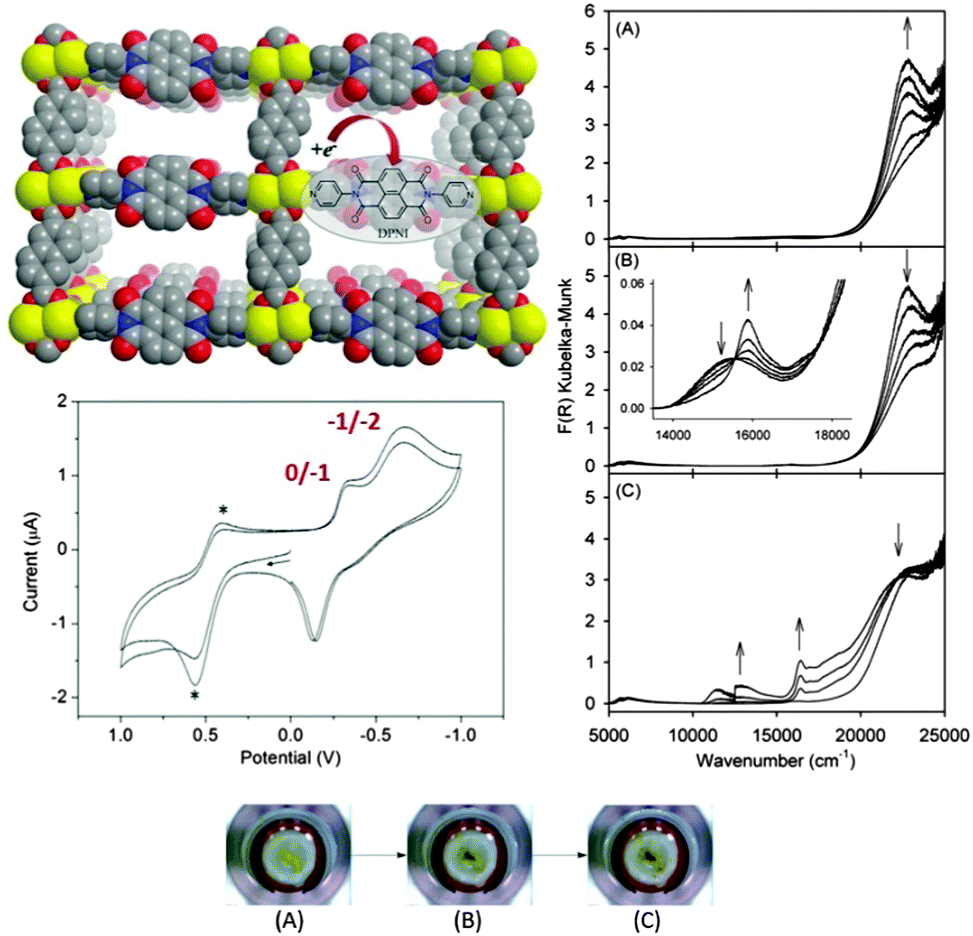 | ||
| Fig. 2 Solid state electrochemistry and vis/NIR spectroelectrochemistry of [Zn2(ndc)2(DPNI)]. Cyclic voltammetry reveals two redox waves corresponding to generation of the radical anion and dianion states of the redox-active DPNI ligand. The optical properties of these states can be rapidly accessed using spectroelectrochemistry for generation of the radical anion (A) and dianion (B) states, which are accompanied by marked colour changes as seen in the photographs of the MOF on the transparent working electrode (bottom). Application of a more reducing potential leads to framework degradation (C). Reproduced from ref. 9 with permission from the Royal Society of Chemistry. | ||
The development of in situ solid state EPR spectroelectrochemistry has enabled the interrogation of the charge transfer MOF [(Zn(DMF))2(TTFTC)(DPNI)], incorporating electron donor TTFTC and acceptor DPNI ligands in a mixed stacking arrangement (Fig. 3).62 The as-synthesised material exhibited the EPR spectrum shown by the black curve in Fig. 3. The splitting of the signal was attributed to two overlapping peaks due to radical species, i.e., a partial CT leads to the coexistence of the TTFTC4− radical cation and DPNI radical anion. At a cathodic potential, the EPR signal due to the TTFTC4− radical cation decreases as the population of DPNI radical anions increases. Coupling the spectral analysis from the EPR spectroelectrochemical reduction with the related UV/vis/NIR measurement proved that partial CT occurred in the as synthesised material. Importantly, this could be reversibly switched on and off by altering the redox state.
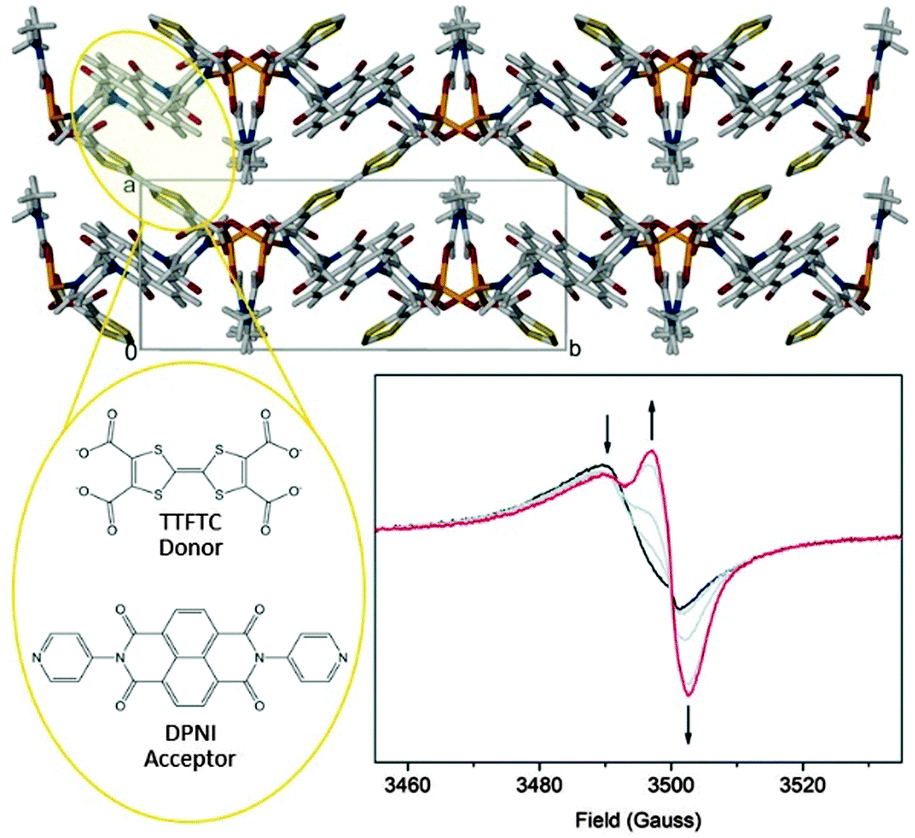 | ||
| Fig. 3 The charge transfer MOF [(Zn(DMF))2(TTFTC)(DPNI)] which exhibits through-space CT interactions. Reduction of the MOF switches the CT off. This is consistent with solid state EPR spectroelectrochemistry which revealed evidence for a concomitant decrease in the TTFTC radical cation, and an increase in the DPNI radical anion signal, thus reducing the donor–acceptor interaction. Reproduced from ref. 62 with permission from the Royal Society of Chemistry. | ||
In situ solid state fluorescence spectroelectrochemistry has also been applied recently to triarylamine-based frameworks,89,90 and may prove to be valuable for assessing the function of materials for optoelectronic device applications.
2.5 Computational approaches to redox activity in MOFs
Significant efforts have been devoted to high throughput screening and modelling of the gas adsorption properties of MOFs for their potential applications in gas separations.91 By comparison, computational studies on the electronic properties of MOFs have been addressed in a relatively limited number of cases.92Modelling redox-active MOFs, particularly those that exhibit significant charge delocalisation, is a challenging task. The extended structures of these materials lead to the introduction of boundary effects for any finite representation. Nevertheless, valuable information has been obtained from density functional theory (DFT) methods by modelling the structures of MOFs as a small number of discrete components in cases where the degree of delocalisation is small. This approach been valuable for modelling the degree of CT in [(Zn(DMF))2(TTFTC)(DPNI)],62 where δ was predicted to be 0.6 (i.e., near the mixed-valence regime discussed in Section 2.2) and in the ferrocene functionalised NU-1000 MOF.93 In the latter case, the Marcus equation was used to describe the rate of intermolecular charge transfer in a chemically relevant fragment from the extended framework crystal structure, and charge transfer was subsequently shown to occur via a superexchange mechanism.38 In addition to modelling the intrinsic charge transfer properties of the MOF, this study also demonstrated the application of computational methods to model MOFs for electrochemical device applications where additional components including the solvent, catalyst and contacts must also be considered.93
In the case of [(Zn(DMF))2(TTFTC)(DPNI)]62 and the ferrocene functionalised NU-1000 MOF,93 the systems lie at the weak coupling limit and involve localised CT in which charge is likely to hop discretely between sites.94 While theories pioneered by Hush95 and Marcus96,97 may be relevant to charge transfer in the localised limit, they are likely to be inadequate for strongly interacting systems where the classical treatment of nuclear motion is not realistic. Note that localised CT theories have been successfully applied to Prussian blue and Berlin green.70,98
Clearly, the development of accurate, low-cost computational methods to describe the electronic properties of MOFs is a highly sought after goal. A key issue is the need for more accurate treatments of dispersion interactions which are important for the description of framework materials and are also not fully accounted for by traditional functionals based on DFT.99 New methods must also address the computationally-intensive nature of time-dependent calculations, and deal with approximations for many electron systems which make them prone to inaccuracy.
In an effort to address these challenges, more advanced techniques are currently under development. For example, the DFT-D3 method provides an empirical correction to DFT to account for long-range dispersion interactions, and has already been successfully used to model gas adsorption in MOFs.100 The Universal Force Field has also recently been extended to deal with transition metal-based MOFs.101 The density functional theory tight-binding (DFTB) method has enabled high-level electronic structure calculations to be obtained for band gaps.102,103 An important strategy for the development of improved high-level computational techniques is the need for tandem experimental studies. Measurements on highly ordered systems including single crystals and surface-grafted MOFs (i.e., SURMOFs) are important in this regard.102
A further point to note is that particular attention has been focused on characterisation and control of band gaps for applications in electronic and optoelectronic devices.26 A significant focus has therefore been on computational predictions of the electronic structure of photochromic MOFs where engineering the band gap is of interest for applications in photocatalysis and solar energy harvesting, amongst others.104
3. Exploiting redox activity in MOFs: recent developments
Modulating the redox states of MOFs provides access to a range of functional properties that may differ from those of the as synthesised material itself, including magnetism, luminescence and host–guest properties which are relevant to ion diffusion in batteries, and molecular sensing, amongst numerous other applications. Manipulating and controlling the redox state of a material requires application of an external stimulus such as electrical potential, chemical oxidation/reduction, light, pressure or electrical field. Since a number of reviews have detailed the potential applications of ionic and charged MOFs10 as well as frameworks in which redox activity is central to the function,18 the emphasis here is on the very latest emerging trends in the field.3.1 Gas separations and storage
Amongst the most prominent potential applications of MOFs are gas storage and separations processes, for which ligand-based redox activity has predominantly been exploited. In all cases, these studies have involved generation of the different redox states via ex situ oxidation or reduction of the materials. In the case of MOFs incorporating redox-active ligands, the result is the creation of a stable radical cation or anion state, and the concomitant incorporation of a counter-ion for charge balance.105Ligand-based redox activity has been exploited in frameworks including [Zn2(ndc)2(DPNI)] and [Zn(ndc)(DBMBI)] where the ligands DPNI and DBMBI exhibit stable radical anion states which are generated by reduction with lithium, sodium or potassium napthalenide (XNp where X = Li+, Na+ or K+).106,107 Gas adsorption studies on the reduced MOFs demonstrated improved uptakes of H2, CO2 and CH4 relative to the neutral parent materials, in addition to increases in the selectivities and isosteric heats of adsorption.108 The origins of these enhancements were suggested to lie in the catenated nature of the materials: introduction of the alkali metal counter cation upon reduction displaced the interpenetrated networks, thus increasing the pore apertures and surface areas.109 The uptake was found to be dependent on the amount of chemical reduction, as well as the ionic radius of the alkali metal salt.110 Beyond an optimum dopant concentration, the adsorbate loading decreased due to pore obstruction by the counterion.
Redox-active MOFs incorporating triarylamine ligands that possess stable radical cation states have also been exploited to fabricate stable metal nanoparticles, and the resulting radical cation impregnated MOFs have demonstrated superior gas adsorption performance relative to their neutral analogues.111,112 An autoredox reaction between an organic triarylamine species incorporated into a framework led to formation of the radical cation of the triarylamine, whilst metal ions such as PdII, AgI and AuIII (amongst others) were reduced to the corresponding Pd, Ag or Au metal nanoparticles. Despite a reduction in surface area for the impregnated MOFs, they demonstrated superior H2 storage performance relative to their neutral counterparts.75
While some controversy exists regarding the mechanisms underlying the enhanced gas sorption in reduced or oxidised MOFs, it is typically acknowledged that the included guest (e.g., the counter-cation in the case of framework reduction and the counter-anion in the case of framework oxidation) represent key adsorption sites, and that these may be more important than adsorption onto the framework itself.113
A further issue relates to how redox activity might be exploited for industrial-scale gas separations processes (e.g., CO2 separation from flue gas)114 given that laboratory experiments performed using ex situ methods do not replicate the practical requirements. One possibility is the use of an electrical swing adsorption (ESA) approach for which monolithic carbons have been used previously.115 This application relies on the Joule effect where the passage of electrical current elicits production of heat which stimulates desorption of adsorbates.116 Clearly, if these redox-active materials are to be employed in applications such as CO2 capture, factors such as the switching response times for sorption/desorption will also be critical, as will the energy demands for desorption.
3.2 Electrochromic devices
A common feature of redox state changes in MOFs is the associated colour changes. Commercial applications of electrochromic devices incorporating redox-active systems such as conductive polymers include car mirrors (e.g., Gentex Corporation's antiglare mirrors), electrochromic sunglasses and smart windows (e.g., adjustable darkening windows in Boeing's 787 Dreamliner).117To date, the most extensively studied framework material for its electrochromic properties is Prussian blue which is readily fabricated as thin films on transparent conductive substrates. Recent work on MOFs has also demonstrated their electrochromic properties,25,41,118 including the potential to exploit postsynthetic metallation of otherwise redox-inactive materials to introduce redox centres of interest for generating electrochromism.119 The application of solid state spectroelectrochemistry to MOFs has also revealed their potential polyelectrochromic properties arising from the distinct colours exhibited by several of their redox-accessible states.62
Specific requirements for the implementation of electrochromic MOFs in practical applications include favourable contrast ratio, colouration efficiency, cycle life and response time. To address these issues, the framework NU-901, a polymorph of NU-1000, was fabricated as a thin film of 1D free-standing nanorods on transparent FTO conducting glass.120 The rapid and reversible electrochromic behaviour observed (yellow to deep blue) was attributed to the nanoscale porosity of the MOF. Specifically, the spatial isolation of the redox-active pyrene units in the MOF led to stabilisation of the intensely coloured radical cations (by preventing their dimerisation) upon one electron oxidation.
3.3 Microporous conductors
Typically, MOFs are electronic insulators, or at best, semiconductors due to the nature of the metal centres and bridging ligands which are predominantly carboxylate-based or unsymmetrical (as in the case of CN−) and do not facilitate strong charge transfer. Of the vast array of crystalline coordination polymers reported to date in the literature, examples of three-dimensional conducting MOFs remain scarce, however, a number of recent studies have made enormous strides in this area.12,43While redox activity alone does not guarantee that a framework material will be intrinsically conducting, effective coupling between redox-active units through overlap of their frontier orbitals is one of the primary means by which conductivity can be achieved. For example, through bond conduction has been suggested to occur through networks of electron-rich heteroatoms such as sulfur in the framework [Mn(dsbdc)].121 A number of semiconducting 2D frameworks based on triphenylene-derived units have been reported, and their applications have been expanded to include chemiresistive sensing of volatile organic compounds.122,123
The opportunity to modulate the redox states of MOFs offers prospects for harnessing switchable conductivity for numerous applications including electrocatalysis and chemiresistive sensing, amongst others.25,27,43,118 Conducting MOFs have been the subject of excellent recent reviews in the field,43,45 and the present focus is on cases where the conducting properties have been modulated as a function of redox state. Clearly, for the increasing number of conductive MOFs reported, consideration of potential redox-modulated properties represents a worthwhile pursuit.
Transition metal dithiolene complexes have been employed extensively as redox-active components of MOFs in light of their electronically-delocalised character50,124–128 which renders them attractive structural motifs for conductive materials. One of the earliest examples of a conductive MOF featuring redox-state dependent conductivity was the dithiolene-based system [Cu{Ni(pdt)2}] which consists of formally NiII centres coordinated by pdt ligands. Chemical oxidation using I2 vapour resulted in a 104-fold increase in conductivity which was attributed to electron hopping between the Ni centres, half of which were oxidised to generate a mixed-valence state. In a more recent example, the 2D framework [Ni3(bht)2] exhibited high charge conduction along the 2D sheets which was facilitated by electron delocalisation through the Ni dithiolene moiety.49,50 The conductivity was also found to be sensitive to the redox-state of the framework.124
A number of MOFs exhibit semiconducting behaviour which arises from charge transport via organic components, particularly those based on the TTF moiety. Recently, a highly porous ZnII framework, [Zn2(TTFTB)] and a series of isostructural analogues based on MnII, CoII and CdII, incorporating a benzoate-functionalised TTF ligand showed both permanent porosity and a high charge mobility that rivalled equivalent organic-based semiconductors.46 Here, the mechanism of conduction was through the TTFTB stacks.46,47 The TTFTB ligand has also been integrated into MOFs in combination with various divalent metal ions and neutral coligands.129 The solid state electrochemical and spectroelectrochemical properties of these materials have demonstrated that two reversible anodic processes corresponding to formation of the radical cation and dication of the TTFTB are accessible. While conductivities for these systems were not reported, the results provided evidence that the properties of the ligand are maintained in the framework.
Redox activity can also be exploited to bring about CT interactions in MOFs which promote long range charge delocalisation and conductivity of the type discussed in Section 2.2. The use of an appropriate D and A pair can induce partial charge transfer which is either through-space62 or through-bond.59,61 In the latter case, infiltration of TCNQ into the three-dimensional framework HKUST-1 enhanced the conductivity by seven orders of magnitude.61 The origin of the enhancement was attributed to postsynthetic coordination of the electron accepting TCNQ moiety to CuII paddlewheel units forming a conductive pathway that spiralled through voids in framework. A three-state superexchange model was invoked to rationalise the new CT features in the absorption spectra,45 implying that simple 2-state models may not be sufficient. Semiclassical theories may therefore prove very valuable, as they have for ligand bridged dinuclear complexes which lie in the intermediate Robin and Day8 Class II–III region for mixed-valence chemistry.74,130 Subsequent computational work provided structural predictions for MOFs in which similar guest-induced emergent properties may be realised.131 This postsynthetic approach to introducing the acceptor moiety is particularly valuable for systematically tuning CT interactions (and hence conductivities) of MOFs to obtain structure–function relationships of the type derived for organic CT salts51 and ruthenium-based MOFs.59
IVCT has also been shown to provide a mechanism for long-range charge delocalisation in the framework [(NBu4)2FeIII2(dhbq)3] (Fig. 4).63 Manipulation of the redox state of the MOF via chemical reduction dramatically decreased the conductivity by reducing the charge carrier mobility. These conductivity changes were also manifested by a reduction in the intensity of the IVCT band, suggesting a close connection between the spectroscopic band parameters and the bulk conductivity.
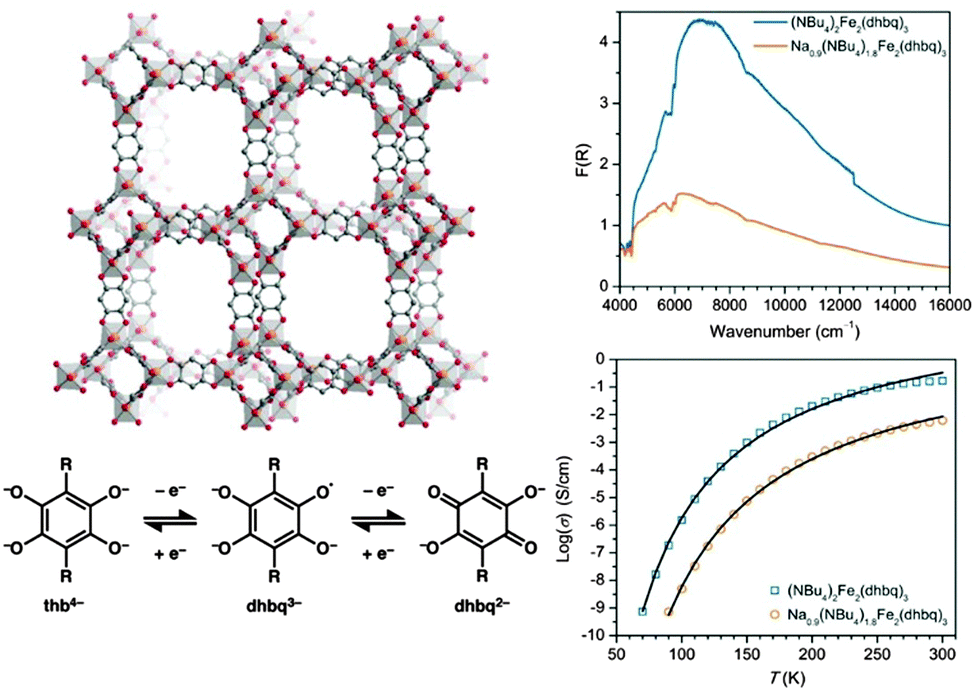 | ||
| Fig. 4 Schematic of the redox-active framework [(NBu4)2FeIII2(dhbq)3] (top left). Chemical reduction generates [(Na)0.9(NBu4)1.8FeIII2(dhbq)3] which is accompanied by a decrease in the intensity of the IVCT band (top right) and a decrease in conductivity (bottom right). These observations are consistent with reduction of the mixed-valence dhbq2−/3− ligands (bottom left). Adapted with permission from ref. 63. Copyright 2015 American Chemical Society. | ||
In addition to spectroscopic methods that provide an indication of new CT interactions within MOFs, measurements of conductivity must ideally be undertaken on single crystal and crystalline films. These techniques are advantageous compared with conventional pressed pellet measurements on coordination solids in order to minimise grain boundary effects and gain insights into anisotropic conduction.12
3.4 Electrocatalysis
While nature has optimised enzymatic processes over the millennia through the use of highly efficient electrocatalytic reactions based on inexpensive, earth-abundant metals such as nickel and iron, the industrial electrocatalysts synthesised today are almost invariably based on expensive and rare platinum-group metals. Spurred by the need for more economically-viable materials based on earth-abundant metals, the development of new classes of electrocatalysts such as MOFs has been gaining momentum owing to their importance in fuel cells for the conversion of chemical energy into electricity132–134 and renewable energy technologies, amongst other areas.135In contrast to existing materials considered for electrocatalysis including metal oxides, porous metal membranes, nanostructured thin films, metal nanoparticles, conducting polymers and discrete transition metal complexes, MOFs are attractive owing to their high surface areas which provide a large concentration of active sites, and their ability to be used in heterogeneous processes.42 Progress in this area has been impeded by the instability of some frameworks to redox cycling, and the intrinsic insulating ability of many MOFs which is not conducive to electron migration through the bulk material. Whilst these issues have been addressed in some part through the fabrication of composites,136 the aforementioned developments in intrinsically conducting MOFs will significantly accelerate progress in this area.
Redox hopping has been shown to provide a mechanism for charge transport within MOFs which have been targeted for a number of electrocatalytic reactions. These processes have largely been focused on key reactions pertinent to artificial photosynthesis including CO2 reduction and water oxidation. Additional electrocatalytic reactions of interest include the hydrogen evolution reaction (HER), oxygen evolution reaction (OER), oxygen reduction reaction (ORR), alcohol oxidation and nitrite reduction amongst others which have been reviewed recently.13,18
A stimulus for the design of MOF-based electrocatalysts has been homogeneous molecular transition metal complexes which are well known for their involvement in particular electrochemical transformations. Inspired by molecular metalloporphyrin electrocatlysts for the reduction of various substrates, a thin film of Fe porphyrin-based [Zr6O4(OH)4(tccp-H2)3] (MOF-525) immobilised on FTO was used to obtain mixture of CO and H2 from the electroreduction of CO2.42 Electrochemical and spectroelectrochemical techniques9 were also employed to elucidate the mechanism of charge transport in an electrocatalytic cobalt framework based on tccp, where a redox hopping mechanism was proposed. The fabrication of thin MOF films for electrocatalysis were shown to be advantageous.39,137
MOFs have also been investigated for water splitting in the context of developing clean energy technologies,138–140 of which OER and HER are key. Polyoxometalate (POM) based MOFs have received significant attention in the latter case where the extensive electrochemical activity of the POMs in electrocatalytic reactions was exploited.13 More recently, a MOF film of NU-1000 was used as a support for deposition of a nickel sulfide electrocatalyst.141 The MOF was shown to improve not only the surface area, but also the proton conductivity of the material.
OER is considered substantially more challenging due to the four-electron nature of this anodic process and the fact that it is performed under alkaline conditions which often promotes framework degradation.139 Nevertheless, O2 formation was observed for frameworks such as analogues of [Zr6O4(OH)4(bpdc)6] (UiO-67), incorporating an Ir-based linker [IrIII(Cp*)-(dcppy)] where O2 was produced. A recent direction in the field is the use of redox-active MOFs as precursors for the preparation of nanoporous carbons and metal oxides.142
An important function of the MOF scaffold is the isolation and subsequent stabilisation of potentially highly reactive catalytic metal centres. In some respects, this is analogous to protective protein structures in nature which stabilise haem with high valent iron-oxo species, which are known to activate strong C–H bonds.143 In this regard, the catalytic conversion of ethane to ethanol was achieved within [Fe2(dobdc)], a redox-active MOF incorporating a coordinatively unsaturated FeII site.143 Upon activation with N2O, the FeII sites were converted to FeIII{hydroxide}, strongly suggesting the presence of an intermediate FeIV{oxo} species from which C–H bonds could potentially be oxidised. When ethane was passed through the framework, various ethane-derived oxygenates such as ethanol and acetaldehyde were formed.
3.5 Energy storage
One of the earliest electronics applications considered for MOFs was their potential for use as rechargeable intercalation electrode materials and electrochemical double layer capacitors relevant to Li-ion battery technologies for portable electronics and fuel cells. Whilst the process of ion conductivity in MOFs has been extensively reviewed,13,18,144 ion diffusion through MOF pores and intrinsic electronic conductivity through the framework backbone are interlinked in many materials.Arguably the first report of a MOF in which the redox activity was exploited in this regard was [FeIII(OH)0.8F0.2(bdc)] (MIL-53(Fe)), which was used as the positive electrode of a Li-ion battery mixed with carbon (Li metal was the negative electrode).64 Lithium ion insertion generated a mixed-valence framework containing FeIII/FeII and Li0/Li+. Whilst the capacity of the material was poor compared with existing devices, it demonstrated the capability of such materials for repeated discharge/recharge cycles. In this case, the lower than expected capacity was attributed to the lack of permanent porosity which restricted Li-ion motion and prevented complete reduction of the FeIII centres, as well as the existence of a redox-inactive ligand (bdc).
MOFs have also been used for negative electrode materials in the context of lithium storage, where the aforementioned issues of capacity were addressed by exploiting the coexistence of redox activities of the metal and ligand sites.18 In the redox-active framework [Cu(aqdc)], in situ XANES and cyclic voltammetry showed that during the discharge process, both the CuII,II2(acetate)4 paddlewheels and anthraquinone groups facilitated the reduction process.145
A number of strategies have now been investigated in an effort to surmount issues with the instability of MOF materials over repeat redox cycles, as well as their poor capacitance and low Faradaic efficiencies due to low electrical conductivities and steric hindrance to Li ion insertion.18 The subject of MOF supercapacitors has been reviewed recently,144 and whilst outside the scope of this article, the majority of approaches involve the destruction of MOFs via pyrolysis, rather than their use as pristine materials. Systems with very high areal capacitance have now been developed by improving the conductivities through doping pristine MOF crystals with graphene146 or polyaniline chains that have been electrochemically deposited on the MOF surface.147 Issues deserving of more attention include the mechanisms of solid state electrochemical behaviour and the electron storage properties which are rarely discussed. The advent of MOFs incorporating redox-active metals and linkers as well as higher electrical conductivities should also instigate further developments in MOFs for energy storage, and coupling of the electronic and ion motion should bring about higher gravimetric capacities.
3.6 Molecular electronics: sensors, switches and machines
An interesting possibility for redox-active MOFs is the prospect of exploiting the switchable nature of the redox states to bring about dynamic motion, as is relevant to molecular machines based on robust and dynamic bistable mechanically interlocked molecules. In this context, the NU-1000 framework incorporating trisradical rotaxanes has recently been addressed (Fig. 5).148 In this proof-of-concept experiment, the MOF scaffold aided the organisation of rotaxanes, which incorporate redox-active viologen subunits that can be addressed electrochemically in the solid state. In particular, the electrochemical reduction resulted in electrochromic behaviour (blue to purple when the radical state is generated, as shown in Fig. 5). This ability to control the electrochromic properties of the material amongst others via a redox-active switch is appealing for the development of solid state molecular electronics devices such as molecular switches and machines.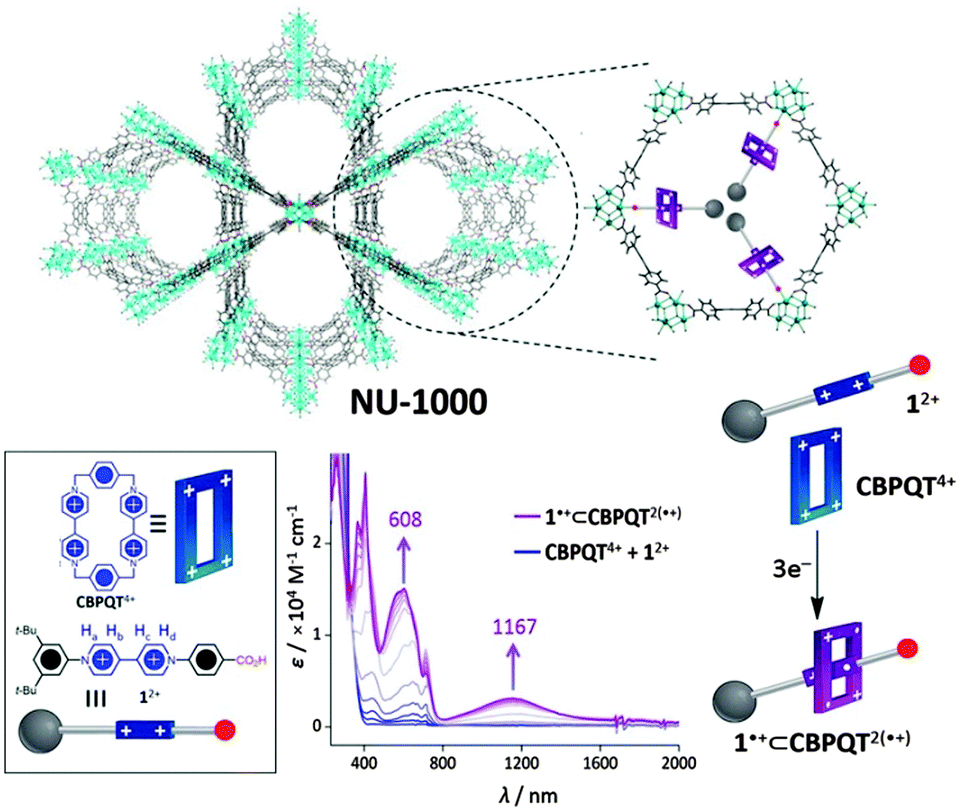 | ||
| Fig. 5 Schematic of NU-1000 containing the semirotaxane (top). Representation of the semirotaxane components 12+ and CBPQT4+ (bottom left). Reduction of 12+ and CBPQT4+ with 3 mol e− generates the semirotaxane 1+⊂CBPQT2(+) (bottom right and centre where the redox-dependent absorption spectrum is shown). Reproduced from ref. 148 with permission from National Academy of Sciences, USA. | ||
The potential to alter the absorption and luminescence properties93,94 of MOFs by exploiting the different characteristics of the redox states is a subject of great interest owing to their potential applications in optoelectronic devices, and as chemical sensors.118,149 For example, a Mn(II) coordination solid based on the tris(4-(pyridin-4-yl)phenyl)amine ligand was found to exhibit switchable ‘on’/‘off’ fluorescence (using chemical oxidation) based on the redox state of the ligand which can exist in its radical cation (‘off’) or neutral state (‘on’).89 Both absorption and fluorescence switching were observed in a redox-switchable MOF that underwent a reversible single-crystal-to-single-crystal transformation.150 In this case, the redox reaction involved the hydroquinone/quinone couple in a hydroxylated analogue of the well-known zirconium UiO-68 framework. The redox triggered transformation exploited a chemical reaction in the crystals, where iodobenzene diacetate was used as the oxidant and ascorbic acid as the reductant.
Different signal transduction methods have been considered for these redox-switchable MOFs, with electrical switching platforms being investigated for integration into devices.118 MOFs exhibiting electrochemiluminescence properties are particularly attractive as their well-ordered porous structures result in high mass transfer capacities which support fast response times. Electrochemiluminescence is appealing due to the low background signal, high sensitivity and wide dynamic range for the detection of metal ions, small molecules and biomacromolecules. The first example involved a ZnII framework which was prepared from a carboxylate modified Ru(2,2′-bpy)2+ moiety, where the RuII guest was electrochemically oxidised to RuIII.151 High electrochemiluminescence emission was observed due to facile electron transfer and was exploited to detect cocaine in serum relevant to illicit drug testing. Hybrid nanocomposites of MOFs with graphene oxide have also proven their utility for sensing acetaminophen and dopamine in serum and urine.152
Recently, the exploitation of redox activity in MOFs for a range of other electroanalytical applications such as nitrite detection has been demonstrated. Here, the development of new sensing devices are relevant to the food and health industries where the use of nitrite as a preserving agent requires close monitoring due to its potential toxicity in excess quantities. A thin film of MOF-525 was constructed from free-base porphyrin linkers and hexa-zirconium nodes on conducting substrates.153 A remarkable current enhancement was detected in the presence of nitrite (NO2−) due to oxidation of NO2− to NO2 by the porphyrin radical cation.
MOFs have also shown utility as alternatives to commercial enzymatic sensors which are currently used for electrochemical hydrogen peroxide (H2O2) and glucose sensing, areas of importance for clinical diagnostics (e.g. for diabetes management) and biotechnology.118 In the presence of H2O2, the CoIII to CoII reduction process in the framework [Co(pbda)(bpy)·2H2O] was sensitive to hydroxyl radicals with an associated colour change and fluorescence response.154 [Cu3(btb)2(H2O)3] (MOF-14) also demonstrated electrocatalytic activity towards H2O2 reduction and glucose oxidation.155
MOF biosensors have recently been developed for the detection of DNA via signal amplification processes. The PCN-222 framework incorporates a redox-active porphyrinic linker which was covalently bound with streptavidin.156 A highly sensitive electrochemical DNA sensor was developed by integrating the electrocatalysis of porphyrinic MOF with a triple-helix molecular switch for signal transduction.156 Hemin has also been encapsulated into Fe-MIL-88 (hemin@MOF) and applied as a redox probe for thrombin with the aid of an enzyme for signal amplification. Hemin is a well-known natural mellaoporphyrin with electroactivity owing to the FeIII/FeII redox couple, but suffers as a catalyst from limited catalytic lifetime. Its incorporation into the host MOF matrix improved its stability, enabling the fabrication of devices from gold nanoparticles functionalised with hemin@MOF.157
3.7 Multifunctionality: the interplay between redox activity and magnetism
Magnetism in MOFs is a relatively well-studied genre.158 The prospect of developing cooperative materials where the magnetic properties can be manipulated via redox state changes has attracted relatively limited attention.The magnetic properties of Hoffman clathrate materials such as [Fe(pyrazine){Pt(CN)4}] change markedly depending on the redox state of the Pt.159 The iodine adduct [Fe(pyrazine){PtII/IV(CN)4(I)n}], prepared by oxidative addition of iodine to the open metal sites of PtII, raised the Curie temperature (Tc) by 100 K. Control of the iodine content (n = 0.0–1.0) resulted in systematic modulation of Tc in the range 300–400 K, showing that the spin transition temperature could be controlled between low and high spin states for magnetically bistable systems.
Systems involving synergistic magnetic and electronic properties are particularly interesting, especially cases where local spins and itinerant electrons interact.160 For the conducting MOF [(NBu4)FeIII2(dhbq)3], the reduced framework [Na0.9(NBu4)1.8FeIII2(dhbq)3] exhibited significantly different conducting and magnetic properties.63 In particular, the magnetic ordering temperature was found to increase in the reduced framework which was a relatively harder magnet due to the additional ligand radical spins which coupled antiferromagnetically to the FeIII centres.
Frameworks based on mixed-valence RuII/III paddlewheels bridged by polycyanide ligands such as TCNQ and its derivatives also exhibit a wide range of redox-dependent properties including antiferromagnetic coupling, long range ferromagnetic ordering and magnetism.161–163
These handful of examples, whilst limited in number, demonstrate the enormous future potential that exists for exploiting the different redox states of MOFs to tune magnetic properties of relevance to the development of new magnetoelectric devices for data storage applications, amongst others.59,63
4. Conclusions
Redox-active MOFs offer the prospect of accessing multiple tunable properties within a given material through manipulation of the redox state. Given the enormous recent strides in the area of conducting frameworks,43,45 routes towards the implementation of these materials at the technological and industrial scales are on the horizon.25,27 The majority of work in the area of redox-active MOFs to date has been focused on bulk synthesis and characterisation, however tremendous advances have been made recently in interfacing MOFs with surfaces, including conductive substrates.20 These methods permit spatial control over the framework film architecture and thus the pore system, enabling the potential anisotropic properties of a framework to be exploited for directional charge transport.At the fundamental level, the development of new computational methodologies to understand the mechanisms of charge transfer in MOFs is gaining momentum. Surface-anchored MOFs have already proven to be particularly relevant in this context.102 Clearly, there is a need for joint experimental, computational and theoretical approaches to explore redox activity. The highly ordered MOF scaffold provides unprecedented opportunities to explore deeply fundamental aspects of charge transfer in three-dimensional coordination space. These understandings promise to shed new light on aspects of charge migration in solar cells, for example, which lie at the heart of renewable clean energy technologies.164
Many significant challenges remain, however, and redox-active MOFs represent a fertile ground for chemists, physicists and engineers to unite their efforts. For the experimental chemist, the ongoing challenge lies in the often serendipitous nature of framework discovery. Nevertheless, principles of ‘crystal engineering’ are instructive in aiding the design of materials.165 A particularly valuable pursuit in this regard is the development of isoreticular series of MOFs which enable systematic modulation of guest species in host MOFs. Ultimately, elucidating structure–function relationships will enable redox-active MOFs to be developed with desired practical functionality. Few systematic studies of this type exist at present; however, if the correct balance of redox-active components can be achieved (in terms of both their electronic and structural characteristics), potentially new fundamental charge transfer phenomena in MOFs may be realised. To date, results in this area suggest that historically important charge transfer theories such as classical and semiclassical theories for organic and inorganic mixed-valence complexes,45 and CT theory for organic donor–acceptor salts,59 may be useful for understanding charge transfer in coordination frameworks.
One caveat to note here is that defects are likely to play a crucial role in dictating the ensuing electronic properties. This issue has been discussed recently and methods are currently under development to control (and exploit) defects.166,167 Significant efforts are required in this area, with combined experimental and computational approaches being a key strategy moving forwards.
Additional challenges and opportunities exist for the experimental chemist including understanding the electrochemistry of porous solids18 and developing methods to modulate redox activity using external stimuli (light, pressure, electrical potential, etc.). While solid state spectroelectrochemical methods have proven to be valuable,9,62 they are problematic when the porosities are low and redox processes are likely to be confined to the surface of MOF particles.
At the applied level, there is significant interest in the integration of redox-active materials into device architectures. Gaining real-time information about the properties of the different redox states (e.g., their optical properties via spectroelectrochemical methods) is valuable here. In particular, it is important to ascertain the structural integrity of a material over repeat redox cycles, especially for practical applications.
Despite a number of crucial challenges for redox-active MOFs at both the fundamental and applied levels, their structural characteristics are highly advantageous. Coupled with new directions in achieving conductivity in these materials, enormous future potential exists to revolutionise a number of fields, particularly those relating to clean energy technologies (fuel cells, batteries, electrocatalysts and solar cells, amongst others).144
List of acronyms for ligands (alphabetical order)
| aqdc | 2,7-Anthraquinonedicarboxylate |
| bdc | 1,4-Benzene dicarboxylate |
| bdc-NH2 | 2-Aminobenzene-1,4-dicarboxylate |
| bht | Benzenehexathiol |
| bpdc | 4,4′-Biphenyl dicarboxylate |
| bpy | 4,4′-Bipyridine |
| 2,2′-bpy | 2,2′-Bipyridine |
| btb | 1,3,5-Benzene tribenzoate |
| btc | 1,3,5-Benzene tricarboxylate |
| CBPQT4+ | Cyclobis(paraquat-p-phenylene) |
| Cp* | Pentamethylcyclopentadienyl |
| DBMBI | N,N′-Di-(4-pyridylmethyl)-1,2,4,5-benzenetetracarboxydiimide |
| DCNQI | N,N′-Dicyanoquinonediimine |
| dcppy | 2-Phenylpyridine-5,4′-dicarboxylate |
| dhbq2−/3− | 2,5-Dioxidobenzoquinone/1,2-dioxido-4,5-semiquinone |
| dobdc | 2,5-Dioxido-1,4-benzenedicarboxylate |
| DPNI | N,N′-Di(4-pyridyl)-1,4,5,8-naphthalenetetracaboxydiimide |
| dsbdc | 2,5-Dithiolatebenzene |
| Fc | Ferrocenyl |
| Fcdc | 1,1′-Ferrocene dicarboxylate |
| hitp | 2,3,6,7,10,11-Hexaiminotriphenylenesemiquinonate |
| H4TBAPy | 1,3,6,8-Tetrakis(p-benzoic acid)pyrene |
| ndc | 2,6-Naphthalene dicarboxylate N,N′-Dicyanoquinodiimine |
| NDI | 1,4,5,8-Napthalene tetracarboxydiimide |
| pbda | 3-(Pyridine-3-yloxy)benzene-1,2-dicarboxylate |
| pdt | Pyrazine-2,3-dithiolate |
| TCNQ | 7,7,8,8-Tetracyanoquinodimethane |
| tcpp | 5,10,15,20-(4-Carboxyphenyl)porphyrin |
| TTF | Tetrathiafulvalene |
| TTFTB | Tetrathiafulvalene tetrabenzoate |
| TTFTC | Tetrathiafulvalene tetracarboxylate |
Acknowledgements
The author gratefully acknowledges the support of a 2015 ChemComm Emerging Investigator Award and the Australian Research Council.Notes and references
- B. F. Hoskins and R. Robson, J. Am. Chem. Soc., 1990, 112, 1546–1554 CrossRef CAS.
- O. M. Yaghi, G. Li and H. Li, Nature, 1995, 378, 703–706 CrossRef CAS.
- S. R. Batten, S. M. Neville and D. R. Turner, Coordination Polymers: Design, Analysis and Application, Royal Society of Chemistry, 2008 Search PubMed.
- S. Kitagawa and R. Matsuda, Coord. Chem. Rev., 2007, 251, 2490–2509 CrossRef CAS.
- A. A. Karyakin, Electroanalysis, 2001, 13, 813–819 CrossRef CAS.
- D. Maspoch, D. Ruiz-Molina and J. Veciana, Chem. Soc. Rev., 2007, 36, 770–818 RSC.
- B. Kong, C. Selomulya, G. F. Zheng and D. Y. Zhao, Chem. Soc. Rev., 2015, 44, 7997–8018 RSC.
- M. B. Robin and P. Day, in Adv. Inorg. Chem. Radiochem., ed. H. J. Emeléus and A. G. Sharpe, Academic Press, 1968, vol. 10, pp. 247–422 Search PubMed.
- P. M. Usov, C. Fabian and D. M. D'Alessandro, Chem. Commun., 2012, 48, 3945–3947 RSC.
- A. Karmakar, A. V. Desai and S. K. Ghosh, Coord. Chem. Rev., 2016, 307, 313–341 CrossRef CAS.
- J. A. Johnson, X. Zhang, X. Zhang and J. Zhang, Curr. Org. Chem., 2014, 18, 1973–2001 CrossRef CAS.
- G. Givaja, P. Amo-Ochoa, C. J. Gomez-Garcia and F. Zamora, Chem. Soc. Rev., 2012, 41, 115–147 RSC.
- A. Morozan and F. Jaouen, Energy Environ. Sci., 2012, 5, 9269–9290 CAS.
- P. Ramaswamy, N. E. Wong and G. K. H. Shimizu, Chem. Soc. Rev., 2014, 43, 5913–5932 RSC.
- S. Horike, D. Umeyama and S. Kitagawa, Acc. Chem. Res., 2013, 46, 2376–2384 CrossRef CAS PubMed.
- H. Al-Kutubi, J. Gascon, E. J. R. Sudholter and L. Rassaei, ChemElectroChem, 2015, 2, 462–474 CrossRef CAS.
- M. Li and M. Dincă, J. Am. Chem. Soc., 2011, 133, 12926–12929 CrossRef CAS PubMed.
- J. E. Halls, D. Jiang, A. D. Burrows, M. A. Kulandainathan and F. Marken, Electrochemistry, 2014, 12, 187–210 CAS.
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933–969 CrossRef CAS PubMed.
- P. Falcaro, R. Ricco, C. M. Doherty, K. Liang, A. J. Hill and M. J. Styles, Chem. Soc. Rev., 2014, 43, 5513–5560 RSC.
- J. L. Zhuang, A. Terfort and C. Wöll, Coord. Chem. Rev., 2016, 307, 391–424 CrossRef CAS.
- O. Shekhah, J. Liu, R. A. Fischer and C. Wöll, Chem. Soc. Rev., 2011, 40, 1081–1106 RSC.
- D. Zacher, O. Shekhah, C. Wöll and R. A. Fischer, Chem. Soc. Rev., 2009, 38, 1418–1429 RSC.
- D. Bradshaw, A. Garai and J. Huo, Chem. Soc. Rev., 2012, 41, 2344–2381 RSC.
- V. Stavila, A. A. Talin and M. D. Allendorf, Chem. Soc. Rev., 2014, 43, 5994–6010 RSC.
- M. D. Allendorf, A. Schwartzberg, V. Stavila and A. A. Talin, Chem. – Eur. J., 2011, 17, 11372–11388 CrossRef CAS PubMed.
- M. D. Allendorf and V. Stavila, CrystEngComm, 2015, 17, 229–246 RSC.
- P. M. Usov, C. McDonnell-Worth, F. Zhou, D. R. MacFarlane and D. M. D'Alessandro, Electrochim. Acta, 2015, 153, 433–438 CrossRef CAS.
- T. B. Faust and D. M. D'Alessandro, RSC Adv., 2014, 4, 17498–17512 RSC.
- M. Meilikhov, K. Yusenko and R. A. Fischer, Dalton Trans., 2010, 39, 10990–10999 RSC.
- J. E. Halls, S. D. Ahn, D. Jiang, L. L. Keenan, A. D. Burrows and F. Marken, J. Electroanal. Chem., 2013, 689, 168–175 CrossRef CAS.
- J. E. Halls, C. Y. Cummings, J. Ellis, L. L. Keenan, D. Jiang, A. D. Burrows and F. Marken, Mol. Cryst. Liq. Cryst., 2012, 554, 12–21 CrossRef CAS.
- C. K. Brozek and M. Dincă, J. Am. Chem. Soc., 2013, 135, 12886–12891 CrossRef CAS PubMed.
- C. K. Brozek, J. T. Miller, S. A. Stoian and M. Dincă, J. Am. Chem. Soc., 2015, 137, 7495–7501 CrossRef CAS PubMed.
- K. Hirai, H. Uehara, S. Kitagawa and S. Furukawa, Dalton Trans., 2012, 41, 3924–3927 RSC.
- J. E. Halls, A. Hernan-Gomez, A. D. Burrows and F. Marken, Dalton Trans., 2012, 41, 1475–1480 RSC.
- M. Meilikhov, K. Yusenko and R. A. Fischer, J. Am. Chem. Soc., 2009, 131, 9644–9645 CrossRef CAS PubMed.
- I. Hod, W. Bury, D. M. Gardner, P. Deria, V. Roznyatovskiy, M. R. Wasielewski, O. K. Farha, J. T. Hupp, W. Bury and O. K. Farha, J. Phys. Chem. Lett., 2015, 6, 586–591 CrossRef CAS PubMed.
- S. R. Ahrenholtz, C. C. Epley and A. J. Morris, J. Am. Chem. Soc., 2014, 136, 2464–2472 CrossRef CAS PubMed.
- I. Hod, W. Bury, D. M. Karlin, P. Deria, C.-W. Kung, M. J. Katz, M. So, B. Klahr, D. Jin, Y.-W. Chung, T. W. Odom, O. K. Farha and J. T. Hupp, Adv. Mater., 2014, 26, 6295–6300 CrossRef CAS PubMed.
- C. R. Wade, M. Li and M. Dincă, Angew. Chem., Int. Ed., 2013, 52, 13377–13381 CrossRef CAS PubMed.
- I. Hod, M. D. Sampson, P. Deria, C. P. Kubiak, O. K. Farha and J. T. Hupp, ACS Catal., 2015, 5, 6302–6309 CrossRef CAS.
- L. Sun, M. G. Campbell and M. Dincă, Angew. Chem., Int. Ed., 2016 DOI:10.1002/anie.201506219.
- D. M. D'Alessandro, J. R. R. Kanga and J. S. Caddy, Aust. J. Chem., 2011, 64, 718–722 CrossRef.
- M. D. Allendorf, M. E. Foster, F. Leonard, V. Stavila, P. L. Feng, F. P. Doty, K. Leong, E. Y. Ma, S. R. Johnston and A. A. Talin, J. Phys. Chem. Lett., 2015, 6, 1182–1195 CrossRef CAS PubMed.
- T. C. Narayan, T. Miyakai, S. Seki and M. Dincă, J. Am. Chem. Soc., 2012, 134, 12932–12935 CrossRef CAS PubMed.
- S. S. Park, E. R. Hontz, L. Sun, C. H. Hendon, A. Walsh, T. Van Voorhis and M. Dincă, J. Am. Chem. Soc., 2015, 137, 1774–1777 CrossRef CAS PubMed.
- A. Aumüller, P. Erk, G. Klebe, S. Hünig, J. U. von Schütz and H.-P. Werner, Angew. Chem., Int. Ed., 1986, 25, 740–741 CrossRef.
- D. Sheberla, L. Sun, M. A. Blood-Forsythe, S. Er, C. R. Wade, C. K. Brozek, A. Aspuru-Guzik and M. Dincă, J. Am. Chem. Soc., 2014, 136, 8859–8862 CrossRef CAS PubMed.
- T. Kambe, R. Sakamoto, K. Hoshiko, K. Takada, M. Miyachi, J.-H. Ryu, S. Sasaki, J. Kim, K. Nakazato, M. Takata and H. Nishihara, J. Am. Chem. Soc., 2013, 135, 2462–2465 CrossRef CAS PubMed.
- G. Saito and Y. Yoshida, Bull. Chem. Soc. Jpn., 2007, 80, 1–137 CrossRef CAS.
- M. R. Bryce, Chem. Soc. Rev., 1991, 20, 355–390 RSC.
- K. P. Goetz, D. Vermeulen, M. E. Payne, C. Kloc, L. E. McNeil and O. D. Jurchescu, J. Mater. Chem. C, 2014, 2, 3065–3076 RSC.
- J. Ferraris, D. O. Cowan, V. Walatka and J. H. Perlstein, J. Am. Chem. Soc., 1973, 95, 948–949 CrossRef CAS.
- J. B. Torrance, Mol. Cryst. Liq. Cryst., 1985, 126, 55–67 CrossRef CAS.
- J. Torrance, J. Vazquez, J. Mayerle and V. Lee, Phys. Rev. Lett., 1981, 46, 253 CrossRef CAS.
- G. Saito and T. Murata, Philos. Trans. R. Soc., A, 2008, 366, 139–150 CrossRef CAS PubMed.
- S. Takaishi, M. Hosoda, T. Kajiwara, H. Miyasaka, M. Yamashita, Y. Nakanishi, Y. Kitagawa, K. Yamaguchi, A. Kobayashi and H. Kitagawa, Inorg. Chem., 2009, 48, 9048 CrossRef CAS PubMed.
- H. Miyasaka, Acc. Chem. Res., 2013, 46, 248–257 CrossRef CAS PubMed.
- K. J. Erickson, F. Leonard, V. Stavila, M. E. Foster, C. D. Spataru, R. E. Jones, B. M. Foley, P. E. Hopkins, M. D. Allendorf and A. A. Talin, Adv. Mater., 2015, 27, 3453–3459 CrossRef CAS PubMed.
- A. A. Talin, A. Centrone, A. C. Ford, M. E. Foster, V. Stavila, P. Haney, R. A. Kinney, V. Szalai, F. El Gabaly, H. P. Yoon, F. Leonard and M. D. Allendorf, Science, 2014, 343, 66–69 CrossRef CAS PubMed.
- C. F. Leong, B. Chan, T. B. Faust and D. M. D'Alessandro, Chem. Sci., 2014, 5, 4724–4728 RSC.
- L. E. Darago, M. L. Aubrey, C. J. Yu, M. I. Gonzalez and J. R. Long, J. Am. Chem. Soc., 2015, 137, 15703–15711 CrossRef CAS PubMed.
- G. Férey, F. Millange, M. Morcrette, C. Serre, M. L. Doublet, J. M. Greneche and J. M. Tarascon, Angew. Chem., Int. Ed., 2007, 46, 3259–3263 CrossRef PubMed.
- H. J. Choi and M. P. Suh, J. Am. Chem. Soc., 2004, 126, 15844–15851 CrossRef CAS PubMed.
- G. C. Allen and N. S. Hush, Prog. Inorg. Chem., John Wiley & Sons, Inc., 1967, pp. 357–389 Search PubMed.
- S. J. England, P. Kathirgamanathan and D. R. Rosseinsky, J. Chem. Soc., Chem. Commun., 1980, 840–841 RSC.
- P. H. Dinolfo, M. E. Williams, C. L. Stern and J. T. Hupp, J. Am. Chem. Soc., 2004, 126, 12989–13001 CrossRef CAS PubMed.
- A. Heckmann and C. Lambert, Angew. Chem., Int. Ed., 2012, 51, 326–392 CrossRef CAS PubMed.
- N. S. Hush, Prog. Inorg. Chem., 1967, 8, 391–444 CrossRef CAS.
- M. D. Ward, Chem. Soc. Rev., 1995, 24, 121–134 RSC.
- D. M. D'Alessandro and F. R. Keene, Chem. Soc. Rev., 2006, 35, 424–440 Search PubMed.
- D. M. D'Alessandro and F. R. Keene, Chem. Rev., 2006, 106, 2270–2298 CrossRef PubMed.
- K. D. Demadis, C. M. Hartshorn and T. J. Meyer, Chem. Rev., 2001, 101, 2655–2685 CrossRef CAS PubMed.
- J. N. Behera, D. M. D'Alessandro, N. Soheilnia and J. R. Long, Chem. Mater., 2009, 21, 1922–1926 CrossRef CAS.
- A. D. Carbó, Electrochemistry of porous materials, CRC press, 2009 Search PubMed.
- A. Domenech, H. Garcia, M. T. Domenech-Carbo and F. Llabres-I-Xamena, J. Phys. Chem. C, 2007, 111, 13701–13711 CAS.
- F. Scholz, L. Nitschke and G. Henrion, Electroanalysis, 1990, 2, 85–87 CrossRef CAS.
- A. M. Bond and F. Scholz, Langmuir, 1991, 7, 3197–3204 CrossRef CAS.
- A. F. Cozzolino, C. K. Brozek, R. D. Palmer, J. Yano, M. Li and M. Dincă, J. Am. Chem. Soc., 2014, 136, 3334–3337 CrossRef CAS PubMed.
- W. Kaim and J. Fiedler, Chem. Soc. Rev., 2009, 38, 3373–3382 RSC.
- L. Dunsch, J. Solid State Electrochem., 2011, 15, 1631–1646 CrossRef CAS.
- F. J. Rizzuto, T. B. Faust, B. Chan, C. Hua, D. M. D'Alessandro and C. J. Kepert, Chem. – Eur. J., 2014, 20, 17597–17605 CrossRef CAS PubMed.
- S. Adeel, M. E. Abdelhamid, A. Nafady, Q. Li, L. L. Martin and A. M. Bond, RSC Adv., 2015, 5, 18384–18390 RSC.
- S. M. Adeel, L. L. Martin and A. M. Bond, J. Solid State Electrochem., 2014, 18, 3287–3298 CrossRef CAS.
- S. V. Bhosale, C. H. Jani and S. J. Langford, Chem. Soc. Rev., 2008, 37, 331–342 RSC.
- E. Coronado, M. Clemente-Leon, J. R. Galan-Mascaros, C. Gimenez-Saiz, C. J. Gomez-Garcia and E. Martinez-Ferrero, J. Chem. Soc., Dalton Trans., 2000, 3955–3961 RSC.
- G. Andric, J. F. Boas, A. M. Bond, G. D. Fallon, K. P. Ghiggino, C. F. Hogan, J. A. Hutchison, M. A. P. Lee, S. J. Langford, J. R. Pilbrow, G. J. Troup and C. P. Woodward, Aust. J. Chem., 2004, 57, 1011–1019 CrossRef CAS.
- C. Hua and D. M. D'Alessandro, CrystEngComm, 2014, 16, 6331–6334 RSC.
- C. Hua, P. Turner and D. M. D'Alessandro, Dalton Trans., 2013, 42, 6310–6313 RSC.
- Y. J. Colon, D. Fairen-Jimenez, C. E. Wilmer and R. Q. Snurr, J. Phys. Chem. C, 2014, 118, 5383–5389 CAS.
- F.-X. Coudert and A. H. Fuchs, Coord. Chem. Rev., 2016, 307, 211–236 CrossRef CAS.
- S. Patwardhan and G. C. Schatz, J. Phys. Chem. C, 2015, 119, 24238–24247 CAS.
- S. Yin, L. Li, Y. Yang and J. R. Reimers, J. Phys. Chem. C, 2012, 116, 14826–14836 CAS.
- N. S. Hush, J. Chem. Phys., 1958, 28, 962–972 CrossRef CAS.
- R. A. Marcus, Rev. Mod. Phys., 1993, 65, 599–610 CrossRef CAS.
- R. A. Marcus, J. Chem. Phys., 1956, 24, 966–978 CrossRef CAS.
- D. M. Pajerowski, T. Watanabe, T. Yamamoto and Y. Einaga, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 153202 CrossRef.
- S. Grimme, Chem. – Eur. J., 2012, 18, 9955–9964 CrossRef CAS PubMed.
- R. B. Getman, Y.-S. Bae, C. E. Wilmer and R. Q. Snurr, Chem. Rev., 2011, 112, 703–723 CrossRef PubMed.
- M. A. Addicoat, N. Vankova, I. F. Akter and T. Heine, J. Chem. Theory Comput., 2014, 10, 880–891 CrossRef CAS PubMed.
- J. Liu, W. Zhou, J. Liu, I. Howard, G. Kilibarda, S. Schlabach, D. Coupry, M. Addicoat, S. Yoneda, Y. Tsutsui, T. Sakurai, S. Seki, Z. Wang, P. Lindemann, E. Redel, T. Heine and C. Woell, Angew. Chem., Int. Ed., 2015, 54, 7441–7445 CrossRef CAS PubMed.
- B. Lukose, B. Supronowicz, P. S. Petkov, J. Frenzel, A. B. Kuc, G. Seifert, G. N. Vayssilov and T. Heine, Condens. Matter, 2011, 1–8 Search PubMed.
- C. H. Hendon, K. E. Wittering, T.-H. Chen, W. Kaveevivitchai, I. Popov, K. T. Butler, C. C. Wilson, D. L. Cruickshank, O. S. Miljanic and A. Walsh, Nano Lett., 2015, 15, 2149–2154 CrossRef CAS PubMed.
- A. Das and D. M. D'Alessandro, CrystEngComm, 2015, 17, 706–718 RSC.
- K. L. Mulfort and J. T. Hupp, J. Am. Chem. Soc., 2007, 129, 9604–9605 CrossRef CAS PubMed.
- C. F. Leong, T. B. Faust, P. Turner, P. M. Usov, C. J. Kepert, R. Babarao, A. W. Thornton and D. M. D'Alessandro, Dalton Trans., 2013, 42, 9831–9839 RSC.
- Y.-S. Bae, B. G. Hauser, O. K. Farha, J. T. Hupp and R. Q. Snurr, Microporous Mesoporous Mater., 2011, 141, 231–235 CrossRef CAS.
- Y.-S. Bae, K. L. Mulfort, H. Frost, P. Ryan, S. Punnathanam, L. J. Broadbelt, J. T. Hupp and R. Q. Snurr, Langmuir, 2008, 24, 8592–8598 CrossRef CAS PubMed.
- K. L. Mulfort, T. M. Wilson, M. R. Wasielewski and J. T. Hupp, Langmuir, 2009, 25, 503–508 CrossRef CAS PubMed.
- M. Meilikhov, K. Yusenko, D. Esken, S. Turner, G. Van Tendeloo and R. A. Fischer, Eur. J. Inorg. Chem., 2010, 3701–3714 CrossRef CAS.
- H. R. Moon, D. W. Lim and M. P. Suh, Chem. Soc. Rev., 2013, 42, 1807–1824 RSC.
- C. Hua, A. Rawal, T. B. Faust, P. D. Southon, R. Babarao, J. M. Hook and D. M. D'Alessandro, J. Mater. Chem. A, 2014, 2, 12466–12474 CAS.
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed.
- C. A. Grande and A. E. Rodrigues, Int. J. Greenhouse Gas Control, 2008, 2, 194–202 CAS.
- A. Subrenat, J. N. Baleo, P. Le Cloirec and P. E. Blanc, Carbon, 2001, 39, 707–716 CrossRef CAS.
- R. J. Mortimer, in Annu. Rev. Mater. Res., ed. D. R. Clarke and P. Fratzl, 2011, vol. 41, pp. 241–268 Search PubMed.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- H. H. Fei, S. Pullen, A. Wagner, S. Ott and S. M. Cohen, Chem. Commun., 2015, 51, 66–69 RSC.
- C. W. Kung, T. C. Wang, J. E. Mondloch, D. Fairen-Jimenez, D. M. Gardner, W. Bury, J. M. Klingsporn, J. C. Barnes, R. Van Duyne, J. F. Stoddart, M. R. Wasielewski, O. K. Farha and J. T. Hupp, Chem. Mater., 2013, 25, 5012–5017 CrossRef CAS.
- L. Sun, T. Miyakai, S. Seki and M. Dincă, J. Am. Chem. Soc., 2013, 135, 8185–8188 CrossRef CAS PubMed.
- M. G. Campbell, D. Sheberla, S. F. Liu, T. M. Swager and M. Dincă, Angew. Chem., Int. Ed., 2015, 54, 4349–4352 CrossRef CAS PubMed.
- M. G. Campbell, S. F. Liu, T. M. Swager and M. Dincă, J. Am. Chem. Soc., 2015, 137, 13780–13783 CrossRef CAS PubMed.
- T. Kambe, R. Sakamoto, T. Kusamoto, T. Pal, N. Fukui, K. Hoshiko, T. Shimojima, Z. F. Wang, T. Hirahara, K. Ishizaka, S. Hasegawa, F. Liu and H. Nishihara, J. Am. Chem. Soc., 2014, 136, 14357–14360 CrossRef CAS PubMed.
- T. B. Faust, P. M. Usov, D. M. D'Alessandro and C. J. Kepert, Chem. Commun., 2014, 50, 12772–12774 RSC.
- J. Cui and Z. Xu, Chem. Commun., 2014, 50, 3986–3988 RSC.
- X.-Y. Li, Y.-G. Sun, P. Huo, M.-Y. Shao, S.-F. Ji, Q.-Y. Zhu and J. Dai, Phys. Chem. Chem. Phys., 2013, 15, 4016–4023 RSC.
- Y. Kobayashi, B. Jacobs, M. D. Allendorf and J. R. Long, Chem. Mater., 2010, 22, 4120–4122 CrossRef CAS.
- B. Chen, Z.-P. Lv, C. F. Leong, Y. Zhao, D. M. D'Alessandro and J.-L. Zuo, Cryst. Growth Des., 2015, 15, 1861–1870 CAS.
- B. S. Brunschwig, C. Creutz and N. Sutin, Chem. Soc. Rev., 2002, 31, 168–184 RSC.
- X. Nie, A. Kulkarni and D. S. Sholl, J. Phys. Chem. Lett., 2015, 6, 1586–1591 CrossRef CAS PubMed.
- W. Xia, A. Mahmood, R. Q. Zou and Q. Xu, Energy Environ. Sci., 2015, 8, 1837–1866 CAS.
- J. K. Sun and Q. Xu, Energy Environ. Sci., 2014, 7, 2071–2100 CAS.
- Y. Q. Ren, G. H. Chia and Z. Q. Gao, Nano Today, 2013, 8, 577–597 CrossRef CAS.
- S. L. Li and Q. Xu, Energy Environ. Sci., 2013, 6, 1656–1683 CAS.
- S. Z. Li and F. W. Huo, Nanoscale, 2015, 7, 7482–7501 RSC.
- C.-W. Kung, T.-H. Chang, L.-Y. Chou, J. T. Hupp, O. K. Farha and K.-C. Ho, Chem. Commun., 2015, 51, 2414–2417 RSC.
- S. B. Wang and X. C. Wang, Small, 2015, 11, 3097–3112 CrossRef CAS PubMed.
- K. Meyer, M. Ranocchiari and J. A. van Bokhoven, Energy Environ. Sci., 2015, 8, 1923–1937 CAS.
- T. Zhang and W. B. Lin, in Metal–Organic Frameworks for Photonics Applications, ed. B. Chen and G. Qian, 2014, vol. 157, pp. 89–104 Search PubMed.
- I. Hod, P. Deria, W. Bury, J. E. Mondloch, C.-W. Kung, M. So, M. D. Sampson, A. W. Peters, C. P. Kubiak, O. K. Farha and J. T. Hupp, Nat. Commun., 2015, 6, 8304 CrossRef CAS PubMed.
- Y. J. Han, J. F. Zhai, L. L. Zhang and S. J. Dong, Nanoscale, 2016, 8, 1033–1039 RSC.
- D. J. Xiao, E. D. Bloch, J. A. Mason, W. L. Queen, M. R. Hudson, N. Planas, J. Borycz, A. L. Dzubak, P. Verma, K. Lee, F. Bonino, V. Crocella, J. Yano, S. Bordiga, D. G. Truhlar, L. Gagliardi, C. M. Brown and J. R. Long, Nat. Chem., 2014, 6, 590–595 CrossRef CAS PubMed.
- L. Wang, Y. Z. Han, X. Feng, J. W. Zhou, P. F. Qi and B. Wang, Coord. Chem. Rev., 2016, 307, 361–381 CrossRef CAS.
- Z. Zhang, H. Yoshikawa and K. Awaga, J. Am. Chem. Soc., 2014, 136, 16112–16115 CrossRef CAS PubMed.
- K. M. Choi, H. M. Jeong, J. H. Park, Y. B. Zhang, J. K. Kang and O. M. Yaghi, ACS Nano, 2014, 8, 7451–7457 CrossRef CAS PubMed.
- L. Wang, X. Feng, L. Ren, Q. Piao, J. Zhong, Y. Wang, H. Li, Y. Chen and B. Wang, J. Am. Chem. Soc., 2015, 137, 4920–4923 CrossRef CAS PubMed.
- P. R. McGonigal, P. Deria, I. Hod, P. Z. Moghadam, A.-J. Avestro, N. E. Horwitz, I. C. Gibbs-Hall, A. K. Blackburn, D. Chen, Y. Y. Botros, M. R. Wasielewski, R. Q. Snurr, J. T. Hupp, O. K. Farha and J. F. Stoddart, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11161–11168 CrossRef CAS PubMed.
- X. J. Zhang, W. J. Wang, Z. J. Hu, G. N. Wang and K. S. Uvdal, Coord. Chem. Rev., 2015, 284, 206–235 CrossRef CAS.
- B. Gui, X. S. Meng, Y. Chen, J. W. Tian, G. L. Liu, C. C. Shen, M. Zeller, D. Q. Yuan and C. Wang, Chem. Mater., 2015, 27, 6426–6431 CrossRef CAS.
- Y. Xu, X. B. Yin, X. W. He and Y. K. Zhang, Biosens. Bioelectron., 2015, 68, 197–203 CrossRef CAS PubMed.
- X. Wang, Q. X. Wang, Q. H. Wang, F. Gao, F. Gao, Y. Z. Yang and H. X. Guo, ACS Appl. Mater. Interfaces, 2014, 6, 11573–11580 CAS.
- C.-W. Kung, T.-H. Chang, L.-Y. Chou, J. T. Hupp, O. K. Farha and K.-C. Ho, Electrochem. Commun., 2015, 58, 51–56 CrossRef CAS.
- L. Z. Yang, C. L. Xu, W. C. Ye and W. S. Liu, Sens. Actuators, B, 2015, 215, 489–496 CrossRef CAS.
- D. Zhang, J. Zhang, R. Zhang, H. Shi, Y. Guo, X. Guo, S. Li and B. Yuan, Talanta, 2015, 144, 1176–1181 CrossRef CAS PubMed.
- P. H. Ling, J. P. Lei and H. X. Ju, Biosens. Bioelectron., 2015, 71, 373–379 CrossRef CAS PubMed.
- S. B. Xie, J. W. Ye, Y. L. Yuan, Y. Q. Chai and R. Yuan, Nanoscale, 2015, 7, 18232–18238 RSC.
- E. Coronado and G. Minguez Espallargas, Chem. Soc. Rev., 2013, 42, 1525–1539 RSC.
- R. Ohtani, K. Yoneda, S. Furukawa, N. Horike, S. Kitagawa, A. B. Gaspar, M. C. Munoz, J. A. Real and M. Ohba, J. Am. Chem. Soc., 2011, 133, 8600–8605 CrossRef CAS PubMed.
- B. Bechlars, D. M. D'Alessandro, D. M. Jenkins, A. T. Iavarone, S. D. Glover, C. P. Kubiak and J. R. Long, Nat. Chem., 2010, 2, 362–368 CrossRef CAS PubMed.
- X. Zhang, M. R. Saber, A. P. Prosvirin, J. H. Reibenspies, L. Sun, M. Ballesteros-Rivas, H. H. Zhao and K. R. Dunbar, Inorg. Chem. Front., 2015, 2, 904–911 RSC.
- Z. Zhang, H. Zhao, M. M. Matsushita, K. Awaga and K. R. Dunbar, J. Mater. Chem. C, 2014, 2, 399–404 RSC.
- Z. Zhang, H. Zhao, H. Kojima, T. Mori and K. R. Dunbar, Chem. – Eur. J., 2013, 19, 3348–3357 CrossRef CAS PubMed.
- W. A. Maza, A. J. Haring, S. R. Ahrenholtz, C. C. Epley, S. Y. Lin and A. J. Morris, Chem. Sci., 2016, 7, 719–727 RSC.
- R. Robson, Dalton Trans., 2008, 5113–5131 RSC.
- Z. Fang, B. Bueken, D. E. De Vos and R. A. Fischer, Angew. Chem., Int. Ed., 2015, 54, 7234–7254 CrossRef CAS PubMed.
- J. Canivet, M. Vandichel and D. Farrusseng, Dalton Trans., 2016, 45, 4090–4099 RSC.
| This journal is © The Royal Society of Chemistry 2016 |