
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlling embedment and surface chemistry of nanoclusters in metal–organic frameworks†
D. E.
Coupry
a,
J.
Butson
b,
P. S.
Petkov
a,
M.
Saunders
c,
K.
O'Donnell
b,
H.
Kim
d,
C.
Buckley
b,
M.
Addicoat
a,
T.
Heine
ae and
P. Á.
Szilágyi
*bf
aDepartment of Physics and Earth Sciences, Jacobs University Bremen, Campus Ring 1, 28759 Bremen, Germany
bDepartment of Physics, Astronomy and Medical Radiation Sciences, Curtin University, GPO Box U1987, Perth, WA 6845, Australia. E-mail: P.A.Szilagyi@greenwich.ac.uk
cCentre for Microscopy, Characterisation and Analysis, M010, Perth, WA 6009, Australia
dNational Institute of Advanced Industrial Science and Technology, Central 5-2, 1-1-1 Higashi, Tsukuba, Ibaraki 305-8565, Japan
eWilhelm-Ostwald-Institut für Physikalische und Theoretische Chemie, Fakultät für Chemie und Mineralogie, Universität Leipzig, Linnéstr. 2, 04103 Leipzig, Germany
fDepartment of Pharmaceutical, Chemical and Environmental Sciences, University of Greenwich, Medway Campus, ME4 4TB Chatham Maritime, UK
First published on 15th March 2016
Abstract
A combined theoretical and experimental approach demonstrates that nanocluster embedment into the pores of metal–organic frameworks (MOF) may be influenced by the chemical functionalisation of the MOF. Furthermore, this results in the surface functionalisation of the embedded nanoclusters, highlighting the potential of MOF scaffolds for the design and synthesis of novel functional materials.
Nanoclusters are ultra-small particles, typically ≤1 nm. Owing to the limited numbers of constituent atoms in this size regime, bulk properties such as the electronic band structure and magnetic domains transform dramatically.1 In addition, these particles exhibit even higher surface-to-volume ratios than conventional nanoparticles, an obvious consequence of their reduced size. This in turn, renders nanoclusters highly desirable for heterogeneous catalysis, which typically only makes use of surface atoms of often costly materials.
Particularly precious metals, such as Au, Pt and Pd, are of great interest as heterogeneous catalysts and their size reduction enables considerable economic benefits.2 Taking into account the dependence of both physical and chemical properties on the cluster size in this regime, it is highly desirable that uniform cluster sizes are obtained. This in turn could also allow for the rational design of metal nanoclusters for purpose.
However, increasing surface-to-volume ratio does not only enhance the clusters' reactivity but it also inseparably results in the increasing surface energies of the nanoclusters.3 Increased surface energies contribute to the thermodynamic destabilisation of nanoclusters. Unstable particles on the other hand stabilise themselves by forming larger clusters, i.e. they sinter. To control nanocluster sizes and shapes one must thus prevent sintering.
In order to stabilise the nanoclusters they need to be supported. There are two fundamentally different approaches to do so, metal nanoclusters may either be capped through colloid chemistry or supported on a scaffold. The former approach has been efficiently applied to precious metal clusters, mainly Au and Pd, and indeed offers reliable size control.4 However, unless the catalytic reaction takes place in the liquid phase, in which the colloid metal nanoclusters may be solubilised, the capped nanoclusters need to be deposited on a 2D support as well. Two-dimensional scaffolds, cf. metal oxides or graphene, supporting either capped or naked nanoclusters, may provide strong interactions with the clusters. However, their reduced dimensionality results in an effectively smaller area available and thus severely impacts the proportion of the overall catalytically active surface area.5
3D scaffolds on the other hand do not display such drawbacks.6 3D porous scaffolds display intrinsically higher surface areas than 2D supports and would enable both liquid and gas-phase reactions without compromising the catalytically active surface area. It is however indispensable that such templates display regular porosity and small and well-defined pore geometries. Furthermore, strong host–guest interactions are necessary to allow the embedding of naked nanoclusters and to render surface capping agents obsolete.
Metal–organic frameworks have been highlighted as promising scaffolds for supporting nano-objects.3a,7 They are a relatively new class of materials built up of inorganic nodes interconnected by organic linkers. They are highly crystalline and feature high and regular porosity including well-defined pore geometries. Furthermore, a broad range of chemical functionality may be introduced in these frameworks through functional groups on the organic linkers.8 In this approach, the MOF pores act as templates for the guest nanoclusters or nanoparticles, depending on their size.
Although these properties underpin how desirable MOF scaffolds are, in real terms they display various issues regarding guest embedment. More specifically, it has been revealed that the embedment of particles in the MOFs' pores is by no means guaranteed. Often the MOF particles have been found to support metal nanoparticles on their outer surfaces rather in their inner pores, or acting as mere 2D supports.7b,9 Despite this obvious drawback, to date no model has been developed to explain this unexpected behaviour.
In this manuscript we show that this phenomenon is due to the discrepancies in the adsorption enthalpy with which the MOF scaffolds bind the various guest materials and that it may be controlled by introducing functionalities in the MOFs and match them with the guest material.
We have selected a robust framework, UiO-66,10 and one of its functionalised analogues, NH2–UiO-66,11 to demonstrate and rationalise this model. Pd was chosen as guest metal for its catalytic relevance and because it has been reported that its embedment in the pristine UiO-66 does not occur.9 On the other hand it has been hypothesised that Pd nanoparticles might be anchored to the functionalised NH2–UiO-66.12 Although it should be noted that in these reports neither the mechanism nor the effect of anchoring are discussed, furthermore, the TEM micrographs provided reveal that the Pd particles were not necessarily embedded in the NH2–UiO-66 pores.13
To verify the suitability of the selected MOFs we developed a protocol based on computational simulations, which were later compared with the experimental data. In the first step, the adsorption of Pd atoms on the selected frameworks has been modelled. All calculations were carried out using the ADF software14 with the PBE-D3 exchange–correlation potential15 and scalar relativity; the TZ2P basis set, a small frozen core and the Becke grid (normal accuracy) for the integration.
The reference UiO-66 cluster was obtained from a cut-out of an experimental crystal structure.10 Hydrogen atoms were added to the structure and their positions were optimised while keeping the heavier atom positions constrained (see Fig. 1). In subsequent calculations, the aromatic rings allowed to freely rotate while the rest of the cluster stayed constrained. For each possible state of amino-functionalisation of the phenyl linkers in both types of cavities, respectively Three-Fold and Four-Fold, several calculations were carried out to describe the inclusion of a single neutral Pd(0) atom, of a Pd(0) dimer and of two distinct Pd(0) atoms in the same cavity. The parameters varying from one calculation to the next were the starting position of the palladium atoms and the orientation of the amine groups. For each case, only the most stable final geometry was retained for further analysis (Fig. 1).
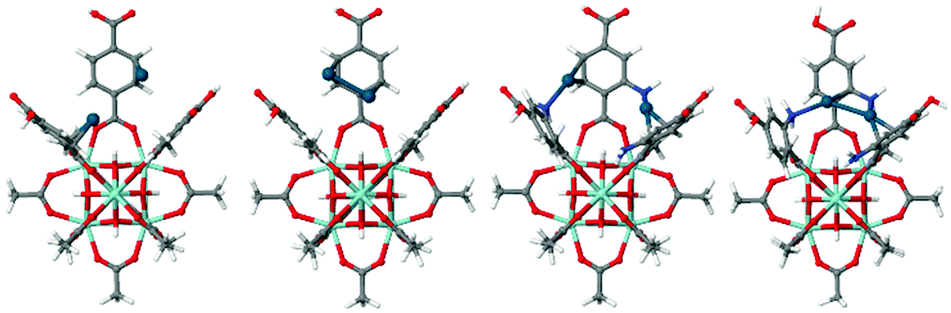 | ||
| Fig. 1 Structure of the model clusters from UiO-66 and NH2–UiO-66 used to calculate the adsorption energy of 2Pd and Pd2 (colour coding: grey – C, red – O, blue – N, cyan – Zr, blue-green – Pd). | ||
The energy difference in the adsorption of Pd as two isolated atoms or a dimer (cluster of two atoms) is related to its tendency to sinter while the adsorption enthalpy of the Pd atoms on the two different matrices reveal the different bond strengths on the pristine and functionalised MOFs.
Table 1 shows that in the case of the functionalised MOF the sintering is less energetically favourable than on the pristine MOFs. This is underpinned by an energy gain of 23 kJ mol−1 from Pd dimerisation on the pristine UiO-66 as opposed to a mere 4 kJ mol−1 (within the error of calculations) on the functionalised NH2–UiO-66. It is also obvious that Pd is more strongly bonded on NH2–UiO-66.
| Modelled adsorption | Adsorption enthalpy (kJ mol−1) | Error (kJ mol−1) | |
|---|---|---|---|
| UiO-66 | Pd, Pd | 150 | 4 |
| Pd2 | 173 | 4 | |
| NH2–UiO-66 | Pd, Pd | 187 | 4 |
| Pd2 | 191 | 4 | |
The strongest adsorption sites were identified according to the simulation as the aromatic ring on the pristine UiO-66 framework, while on the functionalised NH2–UiO-66 it is the NH2 group that is capable of the strongest interactions with Pd.
In order to verify the computational model and check what the difference in the Pd adsorption enthalpies as single atoms or dimers mean for real systems, the model frameworks were synthesised solvothermally, according to Farha et al.16 Given the well-known issues in controlling metal sintering on MOF scaffolds7b a rigorous method has been devised for the addition of Pd. For optimum control of both the amount and location of the Pd precursor the solution-infiltration method has been chosen. Pd was infused from a 7 ml acetonitrile solution of 15 mg Pd(NO3)2 precursor to 100 mg activated MOF and stirred overnight at 40 °C. In addition the Pd precursor was reduced in a hydrogen stream at 210 °C (10 L per hour, 1.3 bar overpressure, 6 hours). This approach offers a much gentler reduction than wet chemical methods such as reduction by sodium borohydride or hydrazine, which often result in the disintegration of the MOF lattice and/or the diffusion of the metal precursor on the MOF particles surface, resulting in the sintering of the reduced atoms.
To compare the effect of MOF linker functionalisation on the embedment of Pd nano-objects High Angle Annular Dark Field-Scanning Transmission Electron Microscopy (HAADF-STEM) micrographs have been obtained (Fig. 2).
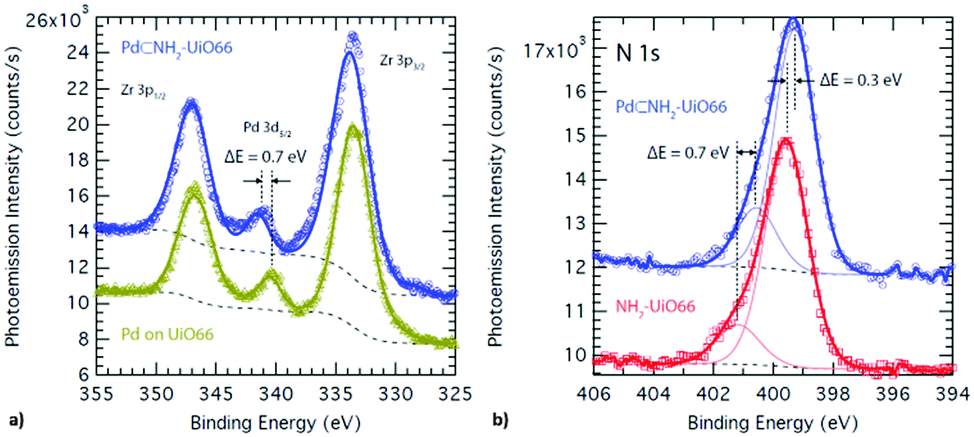 | ||
| Fig. 2 XPS spectrum of (a) Pd supported on UiO-66 (mustard) and embedded in NH2–UiO-66 (blue), and (b) of N in the bare (red) and Pd-supporting (blue) NH2–UiO-66. | ||
Based on their small and uniform particle size, it may be concluded that the Pd nanoclusters were indeed embedded in the pores of the functionalised NH2–UiO-66. In the case of the pristine UiO-66, however, they have sintered on the surface of the MOF particles forming larger, ca. 5–15 nm, nanoparticles. In fact, NH2–UiO-66 has proven to be an excellent scaffold for the embedment of Pd nanoclusters as the size of the Pd nanoclusters, e.g. 0.8 and 1.1 nm, are in very good agreement with that of the MOF's pores, e.g. 0.7 and 1.2 nm.10 This observation is in line with the calculations that highlighted stronger MOF-Pd interactions in the case of the functionalised NH2–UiO-66. Furthermore, this is also underpinned by synchrotron X-ray diffraction data displaying a reduction of NH2–UiO-66 diffraction peak intensities owing to pore filling (ESI†).17
While it is apparent that sintering of Pd atoms could not be avoided as they formed nanoclusters filling the pores of the supporting framework, the MOF-Pd bond was strong enough for the Pd(0) atoms to remain inside the framework's pores and not to diffuse on the particles' surface to form larger and more stable Pd nanoparticles, as in the case of the pristine UiO-66. It should be noted that for an in-depth analysis of the adsorption and sintering of guest particles on a scaffold the enthalpy of metal-atom adsorption with the sublimation enthalpy of the guest are compared by custom. In the case of Pd, the enthalpy of sublimation in the bulk corresponds to 240 kJ mol−1,18 however given the effects on the nanoscale, and below, such as melting point depression there is no useful data available for direct comparison. This highlights the importance of correlating computational and experimental results for the rational embedment of guest materials in the pores of 3D scaffolds.
The difference in the bond strength between the Pd atoms and MOF scaffolds through the effect of functional groups thus resulted in the control of embedment of Pd nanoclusters. In addition to the magnitude of adsorption enthalpy, the binding sites were also revealed to be different for the pristine and functionalised MOFs. X-ray photo-electron spectroscopy (XPS) is sensitive to small changes in the chemical environment and thus is the ideal tool to verify the effect of functionalisation on the surface chemistry of Pd nanoclusters (Fig. 3).
 | ||
| Fig. 3 HAADF-STEM micrographs of (a) Pd nanoparticles on UiO-66 and (b) Pd nanoclusters embedded in NH2–UiO-66. | ||
The Pd 3d3/2 binding energy observed in the XPS spectrum of the Pd supported on the pristine UiO-66 is identical to that observed in the bulk Pd metal and reveals an oxidation state of 0.19 Conversely, the Pd 3d3/2 binding energy observed in the XPS spectrum of the Pd supported on the functionalised NH2–UiO-66 is shifted by ca. +0.4 eV revealing a slight oxidation of the Pd atoms. This however could not amount to the full oxidation-state change of Pd, which would correspond to >2 eV difference. Furthermore, this effect cannot be attributed to a size effect through quantum confinement as XPS probes core electrons and, most importantly, a corresponding reduction shift can also be observed in the binding energy of N 1s when comparing the loaded and unloaded NH2–UiO-66. This is evidence that a bond exists between the surface Pd atoms in the pores and the N atoms of the MOF linkers.
The results reported here highlight for the first time the influence of MOF pore functionalisation on the embedment and surface chemistry of transition metal nanoclusters therein. Considering the interest and potential of nanoclusters as heterogeneous catalysts, whose active atoms are all located on the particle surface, it is anticipated that these findings will have tremendous implications in the design and application of state-of-the-art heterogeneous catalyst nanoclusters.
To the European Commission through the European Research Council (ERC StG C3ENV GA 256962). The authors acknowledge the facilities, and the scientific and technical assistance of the Australian Microscopy & Microanalysis Research Facility at the Centre for Microscopy, Characterisation & Analysis, the University of Western Australia, a facility funded by the University, State and Commonwealth Governments. C. E. B. acknowledges the financial support of the Australia China Science and Research Fund – Joint Research Centre for Energy ACSRF00681. The authors acknowledge the use of equipment, scientific and technical assistance of the WA X-Ray Surface Analysis Facility, funded by the Australian Research Council LIEF grant LE120100026. P. A. S. acknowledges the financial support of the EPSRC, grant number EP/N00938X/1. The synchrotron X-ray experiments were performed at the JAEA beamlines of BL22XU (Proposal No. 2015B3786) at SPring-8 under the Shared Use Program of JAEA Facilities (Proposal No. 2015B-E16). The use of the JAEA beamline of BL22XU is also supported by NIMS microstructural characterisation platform as a programme of “Nanotechnology Platform” of MEXT, Japan under Proposal No. A-15-AE-0017.
Notes and references
- (a) J.-T. Lue, J. Phys. Chem. Solids, 2001, 62, 1599–1612 CrossRef CAS; (b) B. Santiago González, M. J. Rodríguez, C. Blanco, J. Rivas, M. A. López-Quintela and J. M. Gaspar Martinho, Nano Lett., 2010, 10, 4217–4221 CrossRef PubMed; (c) B. Santiago González, M. C. Blanco and M. A. López-Quintela, Nanoscale, 2012, 4, 7632–7635 RSC; (d) Y. Zhu, H. Qian, M. Zhu and R. Jin, Adv. Mater., 2010, 22, 1915–1920 CrossRef CAS PubMed; (e) N. Vilar-Vidal, J. Rivas and M. A. López-Quintela, ACS Catal., 2012, 2, 1693–1697 CrossRef CAS.
- (a) K. L. Kelly, E. Coronado, L. Lin Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668–677 CrossRef CAS; (b) A. A. Herzing, C. J. Kiely, A. F. Carley, P. Landon and G. J. Hutchings, Science, 2008, 319, 1331–1335 CrossRef PubMed; (c) J. Lin, B. Qiao, J. Liu, Y. Huang, A. Wang, L. Li, W. Zhang, L. F. Allard, X. Wang and T. Zhang, Angew. Chem., Int. Ed., 2012, 51, 2920–2924 CrossRef CAS PubMed; (d) M. Turner, V. B. Golovko, O. P. H. Vaughan, P. Abdulkin, A. Berenguer-Murcia, M. S. Tikhov, B. F. G. Johnson and R. M. Lambert, Nature, 2008, 451, 981–983 CrossRef PubMed.
- (a) H. Ri Moon, D.-W. Lim and M. Paik Suh, Chem. Soc. Rev., 2013, 542, 1807–1824 RSC; (b) C. T. Campbell, Acc. Chem. Res., 2013, 46, 1712–1719 CrossRef CAS PubMed; (c) X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025–1102 CrossRef CAS PubMed.
- (a) Z. Huang, X. Gu, Q. Cao, P. Hu, J. Hao, J. Li and X. Tang, Angew. Chem., Int. Ed., 2012, 51, 4198–4203 CrossRef CAS PubMed; (b) A. E. Espinal, L. Zhang, C.-H. Chen, A. Morey, Y. Nie, L. Espinal, B. O. Wells, R. Joesten, M. Aindow and S. L. Suib, Nat. Mater., 2010, 9, 54–59 CrossRef CAS PubMed; (c) C.-H. Chen, L. Jin, A. E. Espinal, B. T. Firliet, L. Xu, M. Aindow, R. Joesten and S. L. Suib, Small, 2010, 6, 988–992 CrossRef CAS PubMed.
- (a) A. M. Argo, J. F. Odzak, F. S. Lai and B. C. Gates, Nature, 2002, 415, 623–626 CrossRef CAS PubMed; (b) O. S. Alexeev, F. Li, M. D. Amiridis and B. C. Gates, J. Phys. Chem. B, 2005, 109, 2338–2349 CrossRef CAS PubMed; (c) A. M. Argo, J. F. Odzak, J. F. Goellner, F. S. Lai, F.-S. Xiao and B. C. Gates, J. Phys. Chem. B, 2006, 110, 1775–1786 CrossRef CAS PubMed; (d) G. N. Vayssilov, B. C. Gates and N. Rösch, Angew. Chem., Int. Ed., 2003, 42, 1391–1394 CrossRef CAS PubMed; (e) G. N. Vayssilov, G. P. Petrova, E. A. Ivanova Shor, V. A. Nasluzov, A. M. Shor, P. St. Petkov and N. Rösch, Phys. Chem. Chem. Phys., 2012, 14, 5879–5890 RSC.
- (a) M. Meilikhov, K. Yusenko, D. Esken, S. Turner, G. Van Tendeloo and R. A. Fischer, Eur. J. Inorg. Chem., 2010, 3701–3714 CrossRef CAS; (b) C. Rösler and R. A. Fischer, CrystEngComm, 2015, 17, 199–217 RSC; (c) J. Juan-Alcañiz, J. Gascon and F. Kapteijn, J. Mater. Chem., 2012, 22, 10102–10118 RSC; (d) P. Á. Szilágyi, E. Callini, A. Anastasopol, C. Kwakernaak, S. Sachdeva, R. van de Krol, H. Geerlings, A. Borgschulte, A. Züttel and B. Dam, Phys. Chem. Chem. Phys., 2014, 16, 5803–5809 RSC; (e) L. Heinke, Z. Gu and C. Wöll, Nat. Commun., 2014, 5, 4562 CAS; (f) W. Guo, J. Liu, P. G. Weidler, J. Liu, T. Neumann, D. Danilov, W. Wenzel, C. Feldmann and C. Wöll, Phys. Chem. Chem. Phys., 2014, 16, 17918–17923 RSC.
- (a) A. Corma, H. García and F. X. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed; (b) H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed; (c) M. Kandiah, S. Usseglio, S. Svelle, U. Olsbye, K. P. Lillerud and M. Tilset, J. Mater. Chem., 2010, 20, 9848–9851 RSC; (d) F. A. Almeida Paz, J. Klinowski, S. M. F. Vilela, J. P. C. Tomé, J. A. S. Cavaleiro and J. Rocha, Chem. Soc. Rev., 2012, 41, 1088–1110 RSC.
- (a) I. Luz, C. Rösler, K. Epp, F. X. Llabrés i Xamena and R. A. Fischer, Eur. J. Inorg. Chem., 2015, 3904–3912 CrossRef CAS; (b) W. Dong, C. Feng, L. Zhang, N. Shang, S. Gao, C. Wang and Z. Wang, Catal. Lett., 2015, 1–9 Search PubMed.
- J. Hafizovic Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef CAS PubMed.
- S. J. Garibay and S. M. Cohen, Chem. Commun., 2010, 46, 7700–7702 RSC.
- X. Li, Z. Guo, C. Xiao, T. Wei Goh, D. Tesfagaber and W. Huang, ACS Catal., 2014, 4, 3490–3497 CrossRef CAS.
- L. Shen, W. Wu, R. Liang, R. Lin and L. Wu, Nanoscale, 2013, 5, 9374–9382 RSC.
- http://www.scm.com, Copyright© 1993-2014: SCM/Vrije Universiteit, Theoretical Chemistry, Amsterdam, The Netherlands.
- (a) S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed; (b) S. Grimme, J. Comput. Chem., 2004, 25, 1463–1473 CrossRef CAS PubMed; (c) J.-M. Ducéré and L. Cavallo, J. Phys. Chem. B, 2007, 111, 13124–13134 CrossRef PubMed.
- M. J. Katz, Z. J. Brown, Y. J. Colón, P. W. Siu, K. A. Scheidt, R. Q. Snurr, J. T. Hupp and O. K. Farha, Chem. Commun., 2013, 49, 9449–9451 RSC.
- (a) N. V. Maksimchuk, M. N. Timofeeva, M. S. Melgunov, A. N. Shmakov, Y. A. Chesalov, D. N. Dybtsev, V. P. Fedin and O. A. Kholdeeva, J. Catal., 2008, 257, 315–323 CrossRef CAS; (b) C. Y. Sun, S. X. Liu, D. D. Liang, K. Z. Shao, Y. H. Ren and Z. M. Su, J. Am. Chem. Soc., 2009, 131, 1883–1888 CrossRef CAS PubMed; (c) Z.-G. Gu, H. Fu, T. Neumann, Z.-X. Xu, W.-Q. Fu, W. Wenzel, L. Zhang, J. Zhang and C. Woll, ACS Nano, 2015, 10, 977–983 CrossRef PubMed; (d) K. Xie, Q. Fu, Y. He, J. Kim, S. J. Goh, E. Nam, G. G. Qiao and P. A. Webley, Chem. Commun., 2015, 51, 15566–15569 RSC.
- Lauro Oliver Paz Barbon, Computational Studies of Transition Metal Alloys, Springer Theses, ch. 6, p. 60 Search PubMed.
- NIST X-ray Photoelectron Spectroscopy Database, Version 4.1 (National Institute of Standards and Technology, Gaithersburg, 2012); http://srdata.nist.gov/xps/.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, HAADF-STEM micrographs, SPXRD patterns and XPS parameters. See DOI: 10.1039/c6cc00659k |
| This journal is © The Royal Society of Chemistry 2016 |