
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Role of highly branched, high molecular weight polymer structures in directing uniform polymer particle formation during nanoprecipitation†
Fiona L.
Hatton
 ,
Pierre
Chambon
,
Alison C.
Savage
and
Steve P.
Rannard
*
,
Pierre
Chambon
,
Alison C.
Savage
and
Steve P.
Rannard
*
Department of Chemistry, University of Liverpool, Crown Street, L69 7ZD, UK. E-mail: srannard@liverpool.ac.uk
First published on 8th February 2016
Abstract
The new macromolecular architecture, hyperbranched polydendrons, are composed of a broad distribution of molecular weights and architectural variation; however, nanoprecipitation of these materials yields highly uniform, dendron-functional nanoparticles. By isolating different fractions of the diverse samples, the key role of the most highly branched structures in directing nucleation and growth has been explored and determined.
Polymeric nanostructures have been formed using various techniques including direct synthesis, such as micro/mini-emulsion polymerisation1,2 and polymerisation induced self-assembly,3–6 or the post-synthesis self-assembly of polymers of varying architectures, hydrophilicity or hydrophobicity.7–9 Alternatively, physical fabrication methods such as the “Particle Replication In Non-wetting Templates” (PRINT™) approach10 may be employed. In many self-assembly routes, a “solvent switch” is used to modify the media environment and drive inter-chain interactions. This can be achieved by changes in pH,11 addition of salt,12 removal of a co-solvent or the slow exchange of good solvent with an anti-solvent13,14 (often water); solution and emulsion approaches have been reported.
A rapid and scale-able solvent switch technique that has gained considerable interest is nanoprecipitation,15 especially in the formation of degradable nanoparticles for biomedical and nanomedicine applications. In general, polymers are dissolved in a good water-miscible solvent and rapidly added to an aqueous anti-solvent. Descriptions of both spontaneous, energy-barrier-free nucleation (spinodal decomposition)16 and nucleation-and-growth mechanisms have been reported. In the latter case, providing the polymer is above its thermodynamic solubility limit within the resulting mixed solution, homogeneous nucleation of particles will occur.16 The particle radii will initially be small, with a free energy of formation that is governed by parameters such as surface tension, particle radius and the free energy difference between media and particle. The free energy will pass through a maximum at a critical nucleus radius leading to rapid growth of larger particles. In either mechanism, growth will continue by depletion of polymer from the surrounding environment or through a process known as diffusion-limited cluster-cluster aggregation, assuming the balance of attractive and repulsive forces will allow interaction and steric/charge stabilisation does not prevent particles from achieving sufficient proximity for effective collision.15 To ensure rapid polymer nucleation, several groups utilise microfluidic or rapid-mixing techniques that allow distinct separation of the nucleation and growth/aggregation stages.17,18
Studies of the impact of polymer architecture and molecular weight on nanoprecipitation outcomes are very rare.15 We have reported a new complex polymer architecture – “hyperbranched polydendrons”19–21 – that overcomes the complexity of dendrimer synthesis by branching between linear–dendritic hybrid polymer chains during propagation (Scheme 1); weight average molecular weights >106 g mol−1, representing >90 conjoined primary linear–dendritic chains, have been achieved. Hyperbranched polydendrons contain considerable variation in: (1) the number of branched chains (Fig. 1), (2) the position of branched points (Fig. 1) and, (3) molecular weight; a significant concentration of un-branched primary chains (linear–dendritic hybrid polymers) is also present. Despite this diversity, the complex mixtures nanoprecipitate to form stable, highly uniform nanoparticle dispersions. Each particle is surrounded by dendron functionality and outcomes are tunable to allow the multi-valency benefits of dendrimers to be achieved at sizes unobtainable by dendrimer synthesis alone (50–200 nm). Hyperbranched polydendrons mimic our studies of branched polymers without dendron chain-ends,22,23 forming more uniform nanoprecipitates with increased stability and reduced size compared to particles generated from their chemically-identical linear primary chain building blocks. As such, they are ideal candidates for detailed examination of the role of the very highly branched, very high molecular weight fraction that is present within the molecular weight distribution.
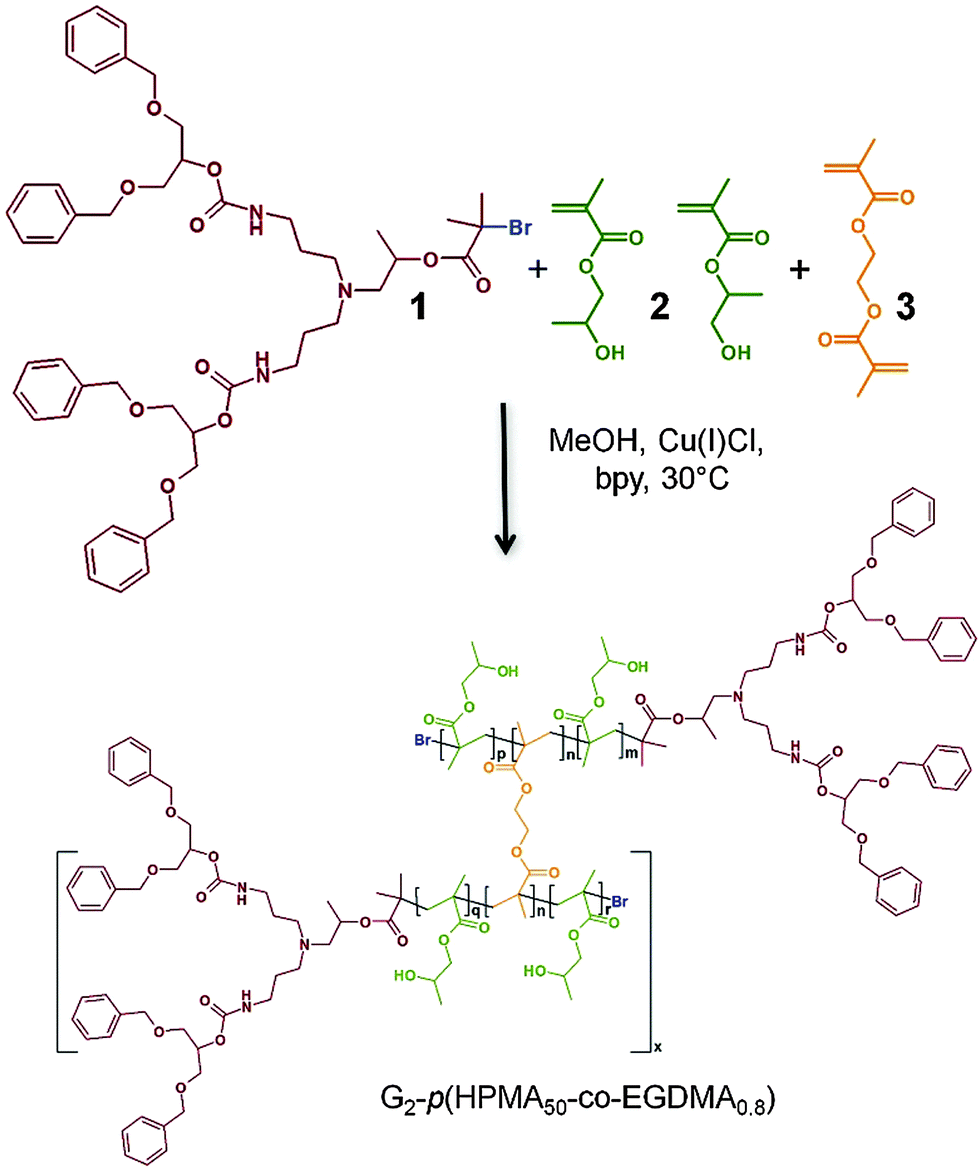 | ||
| Scheme 1 Cu-catalysed ATRP synthesis of hyp-polydendrons using HPMA, 2 (both isomers shown), EGDMA, 3, and a G2 dendritic initiator, 1. | ||
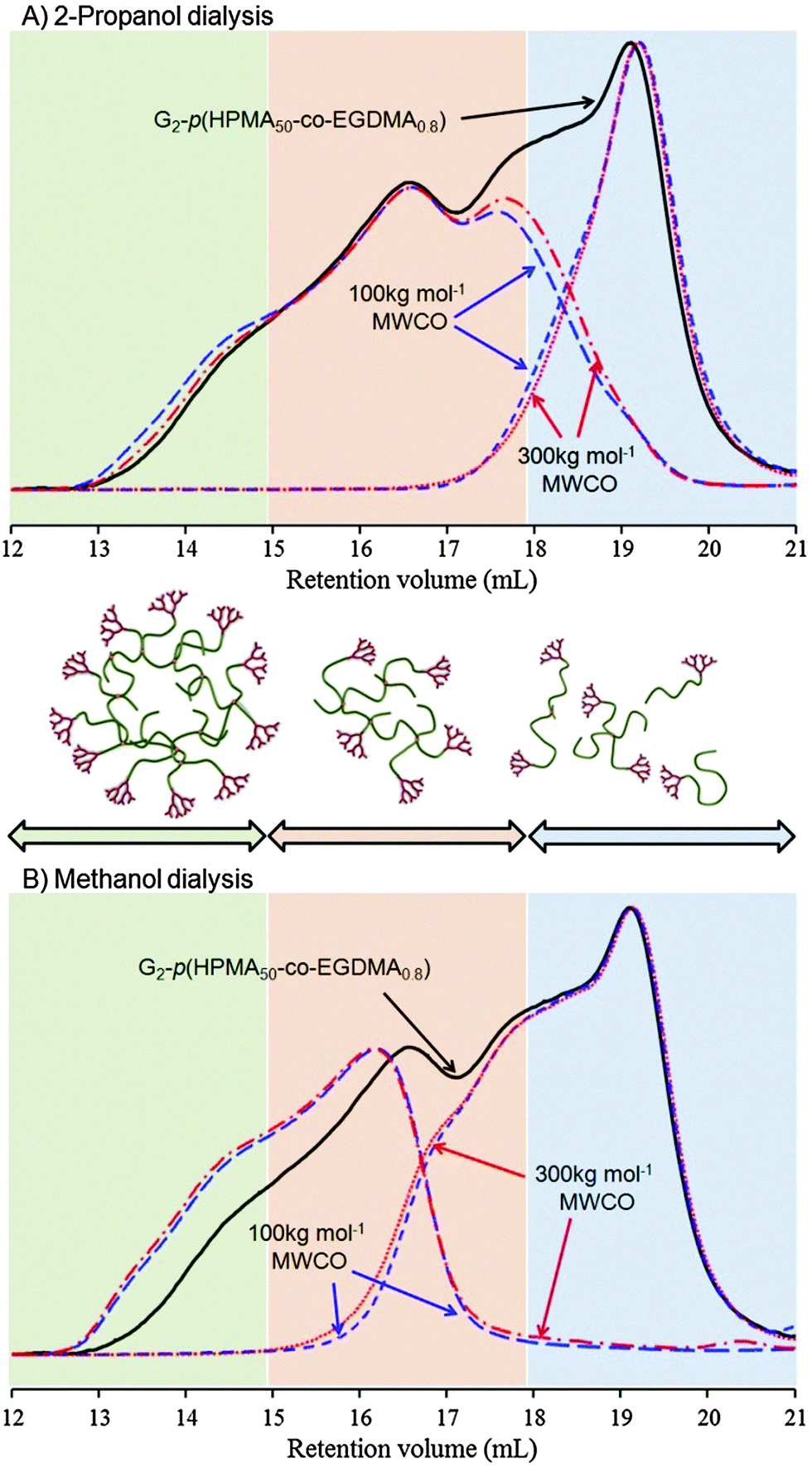 | ||
| Fig. 1 SEC (DMF/0.01 M LiBr eluent) refractive index chromatograms of hyperbranched polydendron samples before (solid black line) and after (red and blue dashed lines) dialysis using either (A) 2-propanol or (B) methanol as the dialysis solvent. Tubing with two different molecular weight cutoffs (MWCO) were utilized in each solvent condition. | ||
Here we isolate the very high molecular weight fraction of hydrophobic hyperbranched polydendrons and study its role in directing successful nanoprecipitation to allow a greater mechanistic understanding and offer more general opportunities for enhanced nanoprecipitation processes.
The polymers were synthesised using previously reported approaches,19–21 namely copper-catalysed methanolic atom transfer radical polymerisation at 30 °C, initiated using a new generation 2 (G2) dendron initiator, 1, and a monomer mixture of 2-hydroxypropyl methacrylate, 2 (HPMA), and ethylene glycol dimethacrylate, 3 (EGDMA) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:3 molar ratio of 1:50:0.8 (target number average degree of polymerisation for each primary chain of 50 monomer units). This ratio also ensures <1 brancher per primary chain and avoids gelation. The number average molecular weight (Mn) and weight average molecular weight (Mw) of the resulting branched polymer (G2-p(HPMA50-co-EGDMA0.8)) were determined by triple detection size exclusion chromatography (DMF/0.01 M LiBr eluent; SEC) to be 115700 g mol−1 and 1538000 g mol−1 respectively (dispersity (Đ) = 13.3; Fig. 1). The equivalent linear polymerisation in the absence of 3 yielded a polymer (G2-p(HPMA50)) of Mn = 12300 g mol−1 and Mw = 15500 g mol−1 (Đ = 1.26). Branching with 3 does not impact the formation of primary chains;24 therefore, it is correct to consider the weight average structures within the G2-p(HPMA50-co-EGDMA0.8) as comprising approximately 100 conjoined G2-p(HPMA50) chains.
:2:3 molar ratio of 1:50:0.8 (target number average degree of polymerisation for each primary chain of 50 monomer units). This ratio also ensures <1 brancher per primary chain and avoids gelation. The number average molecular weight (Mn) and weight average molecular weight (Mw) of the resulting branched polymer (G2-p(HPMA50-co-EGDMA0.8)) were determined by triple detection size exclusion chromatography (DMF/0.01 M LiBr eluent; SEC) to be 115700 g mol−1 and 1538000 g mol−1 respectively (dispersity (Đ) = 13.3; Fig. 1). The equivalent linear polymerisation in the absence of 3 yielded a polymer (G2-p(HPMA50)) of Mn = 12300 g mol−1 and Mw = 15500 g mol−1 (Đ = 1.26). Branching with 3 does not impact the formation of primary chains;24 therefore, it is correct to consider the weight average structures within the G2-p(HPMA50-co-EGDMA0.8) as comprising approximately 100 conjoined G2-p(HPMA50) chains.
This is highly important as Mw represents the molecular weight of macromolecules at the mean of the weight fraction and, therefore, approximately half of the physical mass of the sample comprises branched structures with a minimum of 100 conjoined chains; the Mn value indicates a large number of linear and lesser branched structures, as expected.
G2-p(HPMA50-co-EGDMA0.8) was fractionated using alcoholic dialysis (3 days) with either methanol or 2-propanol, and tubing with two nominal molecular weight cut-off values (MWCO = 100 kg mol−1 and 300 kg mol−1). Two good solvents for p(HPMA) were studied due to the potential for variable wetting and pore swelling within the tubing; outer solutions were collected and fully replenished each day. Solutions within the dialysis tubing, and the combined solvent fractions collected from the reservoir, were dried and the recovered polymer studied by triple detection SEC (Fig. 1 and Table 1).
| Dialysis solvent (tubing MWCO) | Recovery site | G2-p(HPMA50-co-EGDMA0.8) | ||
|---|---|---|---|---|
| M n (SEC, g mol−1) | M w (SEC, g mol−1) | Đ | ||
| a Triple detection SEC using DMF/0.01 M LiBr eluent. b Polymer recovered from solution surrounding dialysis tubing. c Polymer recovered from inside dialysis tubing. See ESI for dn/dc values. | ||||
| — | — | 115700 |
1538000 |
13.3 |
| 2-Propanol | ||||
| 100 kg mol−1 | Externalb | 13300 |
23400 |
1.75 |
| Internalc | 178500 |
1148000 |
6.44 | |
| 300 kg mol−1 | Externalb | 14000 |
21700 |
1.55 |
| Internalc | 136400 |
955800 |
7.01 | |
| Methanol | ||||
| 100 kg mol−1 | Externalb | 28300 |
77500 |
2.74 |
| Internalc | 761200 |
2326000 |
3.06 | |
| 300 kg mol−1 | Externalb | 25400 |
99500 |
3.92 |
| Internalc | 615800 |
1990000 |
3.23 | |
The different MWCO dialysis tubing had a minimal impact on the separation, presumably due to non-aqueous solvent use; SEC chromatograms overlaid almost exactly (Fig. 1) and calculated Mn and Mw values were very similar (Table 1). Methanolic dialysis allowed enhanced isolation of the high molecular weight fraction with the lowest dispersity values.
Nanoprecipitation of the eight fractions (Table 1) and the linear–dendritic hybrid G2-p(HPMA50) was conducted from THF solutions (5 mg mL−1) using deionised water as the anti-solvent and targeting final nanoparticle concentrations of 1 mg mL−1 after THF removal. Dynamic light scattering analysis (DLS; Table 2) of the unfiltered samples yielded z-average diameter (Dz) values of 620–770 nm for nanoprecipitates of G2-p(HPMA50) and the least branched materials fractionated using 2-propanol; similar methanol dialysis fractions generated significant macro-phase separated material. When the high molecular weight fractions isolated using 2-propanol or methanol were nanoprecipitated under identical conditions, considerably more uniform size distributions were observed with Dz values ranging from 135–205 nm and polydispersity (PdI) values ranging from 0.068–0.095 (Table 2).
| Polymer sample (dialysis solvent; tubing MWCO) | Recovery site | D z (nm) | D n (nm) | PdI |
|---|---|---|---|---|
| a DLS measured as unfiltered 1 mg mL−1 aqueous dispersions. b Polymer recovered from solution surrounding dialysis tubing. c Polymer recovered from inside dialysis tubing. d Significant macro-phase separation. | ||||
| G2-p(HPMA50) | — | 670 | 555 | 0.215 |
| Dialysis fractions | ||||
| 2-Propanol | ||||
| 100 kg mol−1 | Externalb | 620 | 525 | 0.214 |
| Internalc | 190 | 165 | 0.070 | |
| 300 kg mol−1 | Externalb | 770 | 390 | 0.424 |
| Internalc | 205 | 175 | 0.095 | |
| Methanol | ||||
| 100 kg mol−1 | Externalb,d | — | — | — |
| Internalc | 145 | 115 | 0.075 | |
| 300 kg mol−1 | Externalb,d | — | — | — |
| Internalc | 135 | 105 | 0.068 | |
To investigate the role of the very high molecular weight species in directing the nanoprecipitation process of highly disperse samples, the high molecular weight fraction of G2-p(HPMA50-co-EGDMA0.8) collected using the 100 kg mol−1 MWCO tubing and methanolic dialysis (G2-p(HPMA50-co-EGDMA0.8)MeOH) was systematically added at varying weight ratios to the G2-p(HPMA50) sample prior to nanoprecipitation. This fraction was selected as it possessed the highest Mn and Mw values and the lowest Đ value of the high molecular weight fractions, thereby allowing the most extreme case of molecular weight and architectural diversity after mixing with G2-p(HPMA50). Nanoprecipitation of a G2-p(HPMA50)/G2-p(HPMA50-co-EGDMA0.8)MeOH mixture containing just 1 wt% branched material generated significantly lower Dz and PdI values than those seen for G2-p(HPMA50) alone; 198 nm (PdI = 0.034) versus 670 nm (PdI = 0.215) respectively (Fig. 2A).
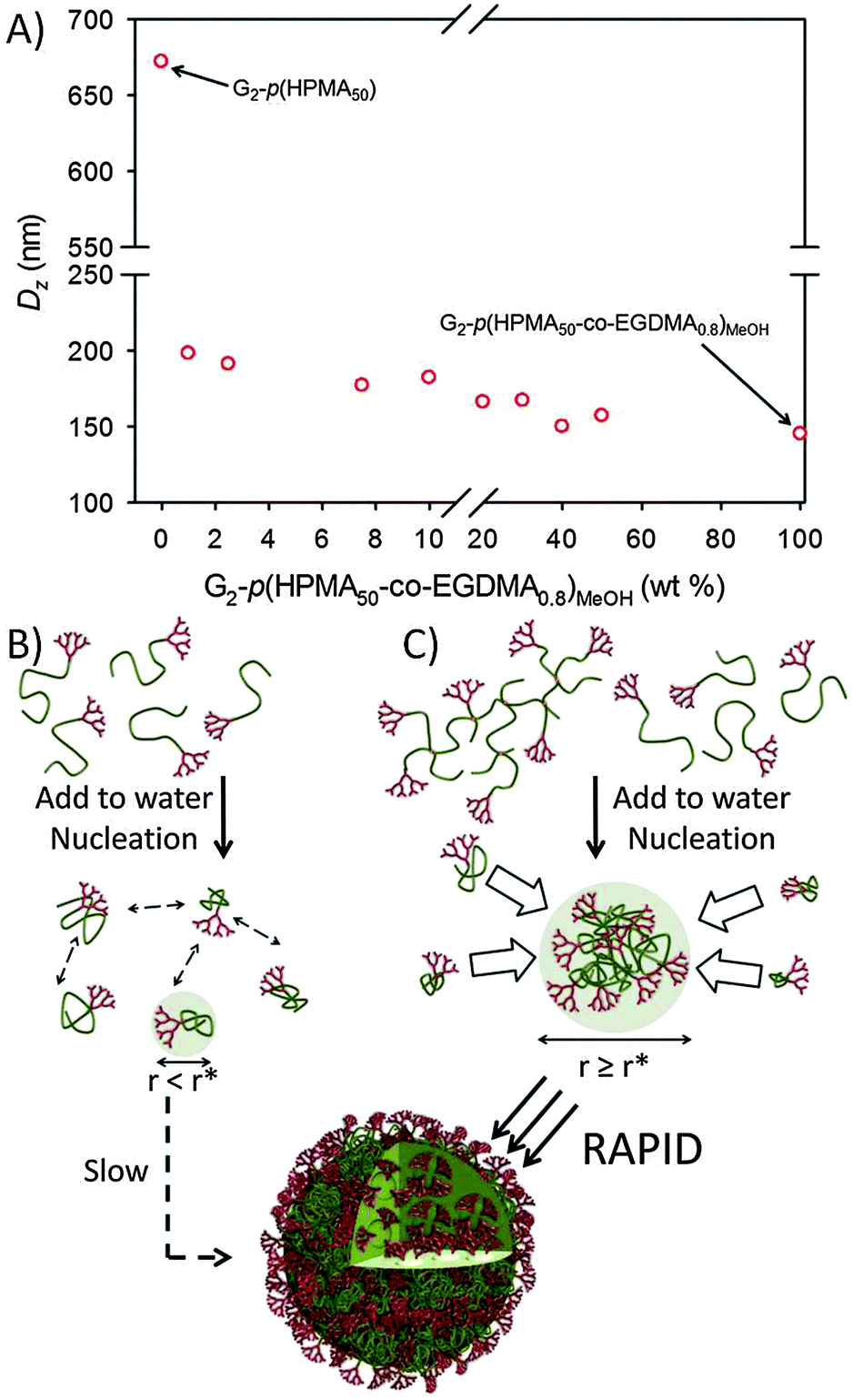 | ||
| Fig. 2 Effect of addition of highly branched, high molecular weight G2-p(HPMA50-co-EGDMA0.8)MeOH to G2-p(HPMA50). (A) Observed modification of nanoprecipitate Dz (DLS) with increasing branched polymer; schematic representation of nucleation from (B) linear–dendritic hybrid polymers with slow growth stage and (C) hyperbranched polydendron generating large nucleus and rapid growth. | ||
Further addition of G2-p(HPMA50-co-EGDMA0.8)MeOH led to a range of nanoparticles with steadily decreasing Dz values (PdI = 0.034–0.110), with the lowest value (145 nm) achieved from nanoprecipitation in the absence of linear G2-p(HPMA50). The impact of highly branched, high molecular weight material within the nanoprecipitation of linear G2-p(HPMA50) can be rationalised by consideration of the nucleation process in the mixed solvent/anti-solvent environment. In the absence of branched polymer, the diffusion of good solvent will lead to the media becoming increasingly less able to solvate the linear–dendritic hybrid polymer chains; these will collapse to form small particles comprising single or small numbers of collapsed macromolecules. It is unlikely that these small nuclei will exceed the critical radius (r*), and slow assembly of chains will lead to poorly defined nucleation and growth stages (Fig. 2B); clearly distinct nucleation and growth are key to the formation of narrow size distributions.15
Conversely, the population of unimolecular structures comprising >100 conjoined chains will lead to the rapid formation of considerably larger nuclei, many of which would be expected to exceed r* and allow a distinct nucleation stage; the linear–dendritic polymer would require a considerable number of single chains to coalesce in a rapid and concerted multi-body collision to achieve the same outcome. We recently reported a lack of discernible light scattering immediately after addition of linear polymer solutions to anti-solvents, in contrast to the observed strong scattering from branched polymers under identical conditions; this would support the absence of rapid nucleation in lower molecular weight linear materials.23 Fractionation to generate G2-p(HPMA50-co-EGDMA0.8)MeOH also increased the number of conjoined chains within the weight average branched structure to >150 (number average structures contain >60 chains) (Table 1).
Decreasing Dz values with increasing content of G2-p(HPMA50-co-EGDMA0.8)MeOH (Fig. 2A) supports a nucleation-driven mechanism as increased nucleation density from additional highly branched polymer will lead to a larger number of smaller nanoparticles at constant polymer mass.25
The number of nanoparticles formed can be approximated by assuming the polymer density and utilising the number average diameter (Dn) values (Table S1, ESI†), and it is important to note that the number of chains and chain ends does not vary throughout this study. Previous studies have shown that increasing polymer molecular weight alone will lead to little difference in Dz values within nanoprecipitations of linear polymer systems, despite considerable variation in solution viscosity, with increased particle sizes resulting for some polymers.26–28 The effects seen here are, therefore, clearly driven by the number of conjoined chains and the architecture of the high molecular weight fraction.
The ability to direct nanoprecipitation outcomes by introducing small fractions of highly branched polymers may have considerable value in ensuring consistency in medical products, decreasing complexity of mixing processes and in controlling nanoparticle sizes and polydispersities. To achieve such results with as little as 1 wt% of branched polymer opens significant opportunities, offers simplification within large scale processes and provides mechanistic insight.
The authors gratefully acknowledge financial support from the University of Liverpool and the Engineering and Physical Sciences Research Council for a PhD studentship (FL) and grant funding (EP/I038721/1) that underpinned the research.
Notes and references
- D. Qi, Z. Cao and U. Ziener, Adv. Colloid Interface Sci., 2014, 211, 47 CrossRef CAS PubMed.
- K. Landfester, A. Musyanovych and V. Mailaender, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 493 CrossRef CAS.
- L. P. D. Ratcliffe, B. E. McKenzie, G. M. D. Le Bouedec, C. N. Williams, S. L. Brown and S. P. Armes, Macromolecules, 2015, 48, 8594 CrossRef CAS.
- R. J. J. Williams, C. E. Hoppe, I. A. Zucchi, H. E. Romeo, I. E. dell'Erba, M. L. Gomez, J. Puig and A. B. Leonardi, J. Colloid Interface Sci., 2014, 431, 223 CrossRef CAS PubMed.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174 CrossRef CAS PubMed.
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Macromolecules, 2012, 45, 6753 CrossRef CAS.
- T. W. Nam, J. W. Jeong, M.-J. Choi, K. M. Baek, J. M. Kim, Y. H. Hur, Y. J. Kim and Y. S. Jung, Macromolecules, 2015, 48, 7938 CrossRef CAS.
- M. S. Onses, L. Wan, X. Liu, N. B. Kiremitler, H. Yilmaz and P. F. Nealey, ACS Macro Lett., 2015, 4, 1356 CrossRef CAS.
- Z. L. Tyrrell, Y. Shen and M. Radosz, Prog. Polym. Sci., 2010, 35, 1128 CrossRef CAS.
- S. E. A. Gratton, P. D. Pohlhaus, J. Lee, J. Guo, M. J. Cho and J. M. DeSimone, J. Controlled Release, 2007, 121, 10 CrossRef CAS PubMed.
- C. Maiti, R. Banerjee, S. Maiti and D. Dhara, Langmuir, 2015, 31, 32 CrossRef CAS PubMed.
- C. Pinto Reis, R. J. Neufeld, A. J. Ribeiro and F. Veiga, J. Nanomed. Nanotechnol., 2006, 2, 8 CrossRef PubMed.
- J. P. Rao and K. E. Geckeler, Prog. Polym. Sci., 2011, 36, 887 CrossRef CAS.
- J. Jiang, Y. Liu, Y. Gong, Q. Shu, M. Yin, X. Liu and M. Chen, Chem. Commun., 2012, 48, 10883 RSC.
- E. Lepeltier, C. Bourgaux and P. Couvreur, Adv. Drug Delivery Rev., 2014, 71, 86 CrossRef CAS PubMed.
- D. Horn and J. Rieger, Angew. Chem., Int. Ed., 2001, 40, 4330 CrossRef CAS.
- N. Anton, F. Bally, C. A. Serra, A. Ali, Y. Arntz, Y. Mely, M. Zhao, E. Marchioni, A. Jakhmola and T. F. Vandamme, Soft Matter, 2012, 8, 10628 RSC.
- K. M. Pustulka, A. R. Wohl, H. S. Lee, A. R. Michel, J. Han, T. R. Hoye, A. V. McCormick, J. Panyam and C. W. Macosko, Mol. Pharmacol., 2013, 10, 4367 CrossRef CAS PubMed.
- F. L. Hatton, P. Chambon, T. O. McDonald, A. Owen and S. P. Rannard, Chem. Sci., 2014, 5, 1844 RSC.
- F. L. Hatton, L. M. Tatham, L. R. Tidbury, P. Chambon, T. He, A. Owen and S. P. Rannard, Chem. Sci., 2015, 6, 326 RSC.
- H. E. Rogers, P. Chambon, S. E. R. Auty, F. Y. Hern, A. Owen and S. P. Rannard, Soft Matter, 2015, 11, 7005 RSC.
- R. A. Slater, T. O. McDonald, D. J. Adams, E. R. Draper, J. V. M. Weaver and S. P. Rannard, Soft Matter, 2012, 8, 9816 RSC.
- J. Ford, P. Chambon, J. North, F. L. Hatton, M. Giardiello, A. Owen and S. P. Rannard, Macromolecules, 2015, 48, 1883 CrossRef CAS.
- J. Rosselgong, S. P. Armes, W. R. S. Barton and D. Price, Macromolecules, 2010, 43, 2145 CrossRef CAS.
- Q. Wang, S. Samitsu, Y. Fujii, C. Yoshikawa, T. Miyazaki, H. Banno and I. Ichinose, J. Polym. Sci., Part B: Polym. Phys., 2015, 53, 615 CrossRef CAS.
- P. Legrand, S. Lesieur, A. Bochot, R. Gref, W. Raatjes, G. Barratt and C. Vauthier, Int. J. Pharm., 2007, 344, 33 CrossRef CAS PubMed.
- M. Beck-Broichsitter, E. Rytting, T. Lebhardt, X. Wang and T. Kissel, Eur. J. Pharm. Sci., 2010, 41, 244 CrossRef CAS PubMed.
- R. Stepanyan, J. G. J. L. Lebouille, J. J. M. Slot, R. Tuinier and M. A. Cohen Stuart, Phys. Rev. Lett., 2012, 109, 138301 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details, NMR spectra, GPC chromatograms, SEM images of nanodispersions, graphs of nanoprecipitate characterisation. See DOI: 10.1039/c6cc00611f |
| This journal is © The Royal Society of Chemistry 2016 |