
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phosphinosilylenes as a novel ligand system for heterobimetallic complexes†
Nora C.
Breit
a,
Carsten
Eisenhut
a and
Shigeyoshi
Inoue
*ab
aTechnische Universität Berlin, Anorganische Chemie, Straße des 17. Juni 135, Sekr. C2, D-10623 Berlin, Germany
bTechnische Universität München, Institute of Silicon Chemistry and Catalysis Research Center, Lichtenbergstraße 4, D-85748 Garching, Germany. E-mail: s.inoue@tum.de
First published on 1st March 2016
Abstract
A dihydrophosphinosilylene iron complex [LSi{Fe(CO)4}PH2] has been prepared and utilized in the synthesis of novel heterobimetallic complexes. The phosphine moiety in this phosphinosilylene complex allows coordination towards tungsten leading to the iron–tungsten heterobimetallic complex [LSi{Fe(CO)4}PH2{W(CO)5}]. In contrast, the reaction of [LSi{Fe(CO)4}PH2] with ethylenebis(triphenylphosphine)platinum(0) results in the formation of the iron–platinum heterobimetallic complex [LSi{Fe(CO)4}PH{PtH(PPh3)2}] via oxidative addition.
Heterobimetallic complexes have received a great deal of attention and have become important targets since they can enhance catalysis through cooperativity.1 N-heterocyclic carbenes are important compounds as supporting ligands in catalysis and for reactive species.2,3 Several heterobimetallic species containing one or more carbenes have been reported in the last decade.1b,4 One inherent problem for the synthesis of heterobimetallic bis(carbene)s (A, Chart 1) is the selectivity of a first monometallation.1b,4a–e Incorporating different ligands like phosphines (B, Chart 1) can facilitate selective product formation.4f,g Another very recent example of utilizing different donors is the silylene–carbene monometallic complex (C, Chart 1).5 Various silylene transition metal complexes have been reported to date and some of those have been tested in catalysis and showed very promising results.6 Also, about a dozen bis(silylene) complexes have been reported to date.7 In most of them the bis(silylene)s are acting as chelating ligands (D, Chart 1)8 with a few exceptions of homobimetallic species (E, Chart 1).8c,9 It should be noted that also few, very interesting heterobimetallic silylene complexes have been described.8b,f,10 However, to the best of our knowledge, no general route for a systematic synthesis of heterobimetallic complexes with a silylene and an additional donor has been reported to date.
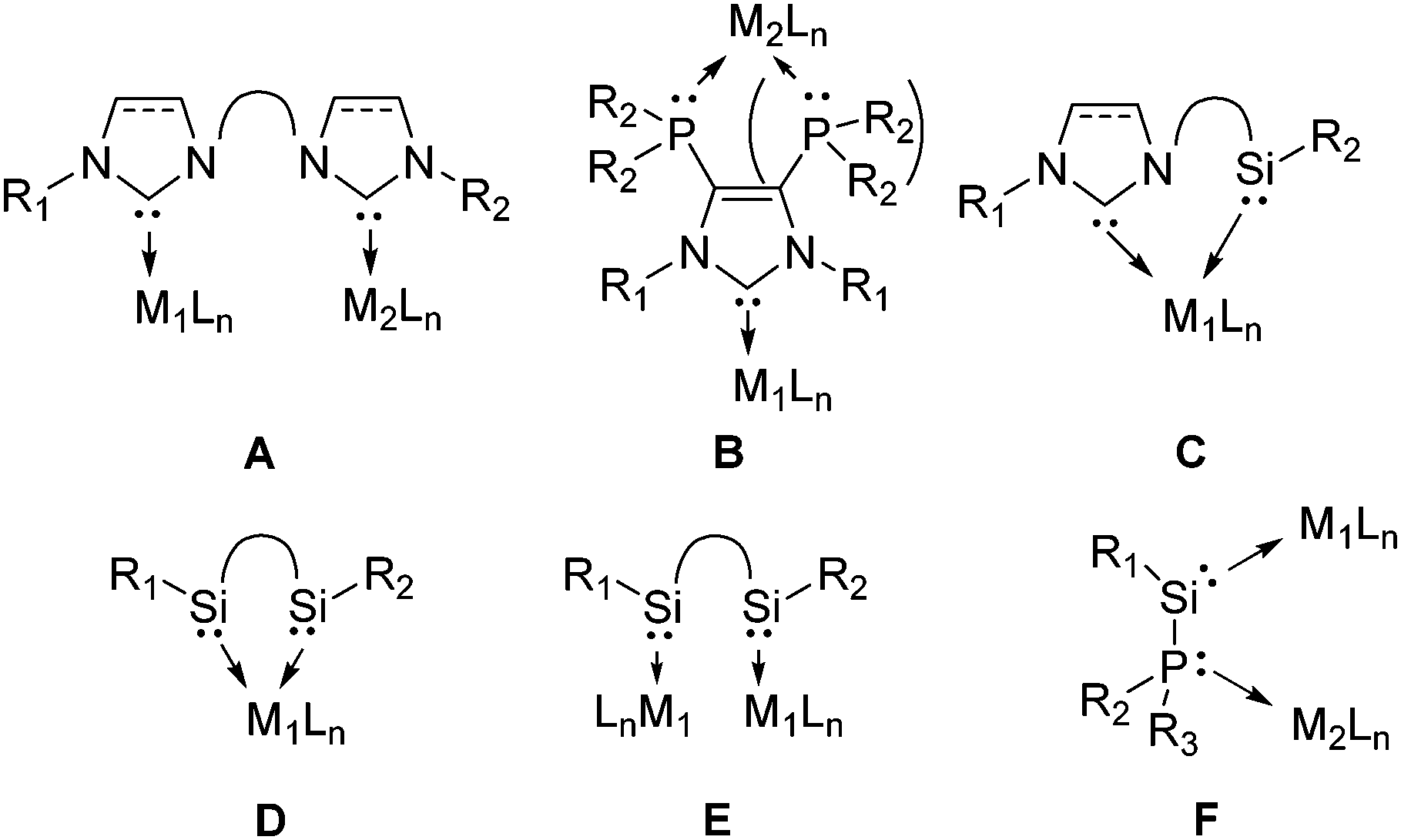 | ||
| Chart 1 Concept of phosphinosilylenes for heterobimetallic complexes. | ||
In this publication we propose a new ligand system for heterobimetallic complexes with interconnected silylene and phosphine donors, namely the phosphinosilylene (F, Chart 1). Only a few stable phosphinosilylenes have been known to date11 and their reactivity studies are rather limited.8c,12
We lay out the systematic synthesis of phosphinosilylene heterobimetallic complexes (F, Chart 1) based on phoshinosilylene 1 (Scheme 1).11c In a stepwise fashion, first the stronger silylene donor should be coordinated to one metal center. Afterwards, the phosphine will be coordinated to the second metal center. In this context, the phosphinosilylene iron carbonyl complex [LSi{Fe(CO)4}P(SiMe3)2] (2) [L = PhC(NtBu)2−] was synthesized by the reaction of 1 with [Fe(CO)5] in a good yield of 78% (Scheme 1). The 29Si{1H} NMR spectrum of 2 shows a significant downfield shift from starting material 111c as well as a downfield shift from the related tungsten complex [LSi{W(CO)5}P(SiMe3)2] (δ = 102.6 ppm, δ = 44.0 ppm,11c and δ = 70.7 ppm,12c respectively) due to a stronger coordination of the silylene to iron in comparison with tungsten, which was similarly reported for other Si(II) compounds.13 The 31P{1H} NMR chemical shifts of these compounds display less variation (δ = −194.4 ppm, δ = −211.0 ppm11c) in agreement with the coordination of the silylene. The presence of the iron carbonyl group is also confirmed by 13C{1H} NMR spectroscopy displaying a signal at δ = 217.2 ppm, which is only slightly downfield shifted from that of [LSi{Fe(CO)4}OtBu] (δ = 216.7 ppm).14 The IR bands of 2 (νCO = 2022, 1941, 1904 cm−1) are close to those of [LSi{Fe(CO)4}OtBu] (νCO = 2026, 1949, 1899 cm−1).14
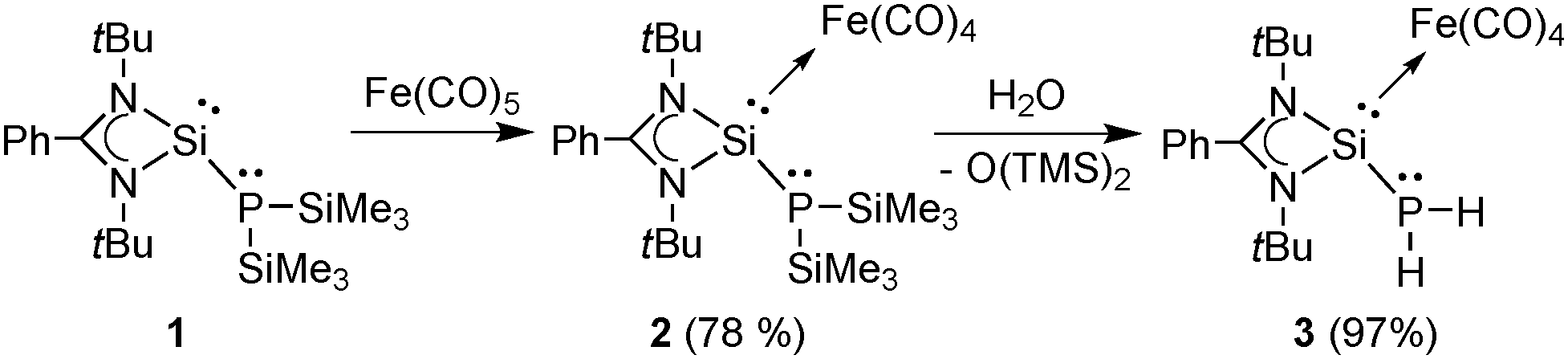 | ||
| Scheme 1 Syntheses of the phosphinosilylene iron carbonyl complexes 2 and 3. | ||
Unfortunately, the phosphinosilylene iron complex 2 does not undergo a reaction with different transition metal complexes ([Fe2(CO)9], [W(CO)5·thf], [Ni(COD)2]) presumably due to the steric bulk of the trimethylsilyl groups. We were able to circumvent this issue by replacing the trimethylsilyl groups with hydrogens. Little attention was yet given to the reaction of RP(SiMe3)2 and H2O yielding RPH2 and O(SiMe3)2.15 However, it proved to be a viable route for the synthesis of [LSi{Fe(CO)4}PH2], 3 (Scheme 1). The formation of 3 is quantitative (97% yield) and little excess water seems not to harm this reaction and product. The signals of the PH2 group in the 1H and 31P NMR spectra appear as a doublet at δ = 2.50 ppm and a triplet at δ = −198.6 ppm with a coupling constant of 1JP–H = 188 Hz. The 29Si{1H} NMR spectrum of 3 reveals a downfield shift from 2 (δ = 112.8 ppm and δ = 102.6 ppm, respectively). The CO signal was found at δ = 216.1 ppm in the 13C{1H} NMR spectrum. In the IR spectra a slight change of the νCO-bands to higher wave numbers was observed (νCO: 2025, 1946, 1913 cm−1 (3) and νCO: 2022, 1941, 1904 cm−1 (2)).
The structural features of compounds 2 and 3 (Fig. 1) are very similar to those of its tungsten analogue [LSi{W(CO)5}P(SiMe3)2].12c The Si1–P1 bond lengths of 2 and 3 (2.2281(6) Å and 2.2551(9) Å, respectively) are shortened compared to 1 (2.2838(12) Å11c). Their iron–silicon bond lengths (2.2777(5) Å in 2 and 2.2412(7) Å in 3) are longer than that of [LSi{Fe(CO)4}OtBu] (2.237(7) Å),14 but shorter than in the bisamidinato species [(PhC{NiPr}2)2Si{Fe(CO)4}] (2.3175(6) Å).13a
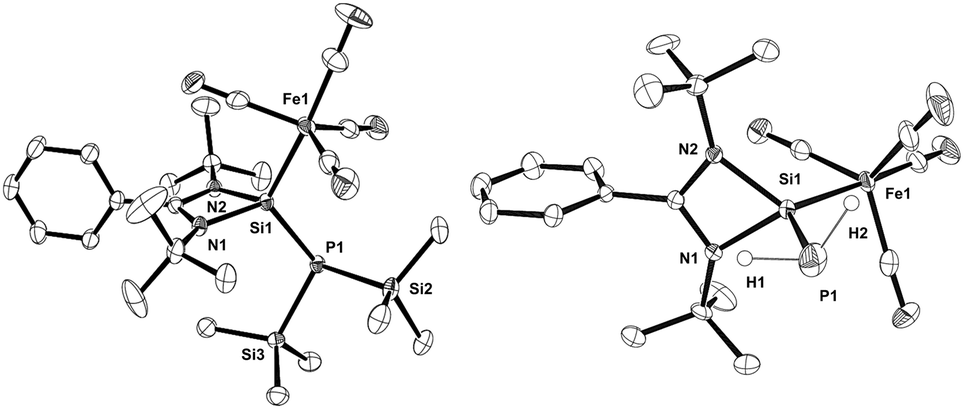 | ||
| Fig. 1 Molecular structures of compounds 2 (left) and 3 (right). Thermal ellipsoids are drawn at the 40% probability level. One disordered tBu group in 2 and hydrogen atoms except for H1 and H2 in 3 are omitted for clarity. Selected bond lengths (Å) and angles (°) in 2 and 3 (for 3 see the values in brackets): P1–Si1 2.2281(6) {2.2551(9)}, Fe1–Si1 2.2777(5) {2.2412(7)}, P1–Si1–Fe1 119.19(2) {119.97(3)}. | ||
With the less sterically crowded compound 3 at hand, we were able to successfully synthesize the first heterobimetallic phosphinosilylene complex 4 (Scheme 2). The phosphine of 3 easily coordinates to pentacarbonyl tungsten and 4 was formed as the major product. This coordination is evident from 1H NMR spectroscopy revealing the shifted PH2 signal with the expected increased coupling constant (1JP–H = 302 Hz in 4 and 1JP–H = 188 Hz in 3; compare also [PH2(SiMe3){W(CO)5}] with 1JP–H = 310 Hz16). In addition, the 31P{1H} NMR signal at δ = −165.2 ppm exhibits tungsten satellites with a coupling constant of 1JW–P = 187 Hz. This coupling constant lies in between those of [PH3{W(CO)5}] and [P(SiMe3)3{W(CO)5}] (1JW–P = 216 Hz and 1JW–P = 150 Hz, respectively).16 The two carbonyl signals appear in the 13C{1H} NMR spectrum at δ = 196.4 ppm (W(CO)5) and δ = 215.1 ppm (Fe(CO)4). The 29Si{1H} NMR signal of 4 is shifted upfield from 3 and is exhibiting a smaller silicon–phosphorus coupling constant (δ = 98.0 ppm, 1JSi–P = 24 Hz and δ = 112.8 ppm, 1JSi–P = 62 Hz, respectively). This suggests that the coordination of the phosphine to tungsten is having a considerable effect on the silicon–phosphorus bond. Despite our best efforts, the isolation of analytically pure 4 was not possible. The solid obtained in 60% yield contains little impurities of W(CO)6 or other W(CO)x byproducts that could not be separated.
 | ||
| Scheme 2 Syntheses of the hetero- and homobimetallic phosphinosilylene complexes 4, 5 and 6. | ||
The molecular structure of 4 was unequivocally assigned by X-ray diffraction analysis (Fig. 2). The iron and tungsten carbonyl moieties are pointing in opposite directions (torsion angle Fe1–Si1–P1–W1: −172.35(4)°) and their interatomic distance amounts to 6.3071(5) Å, which is expected due to their steric requirements. The silicon–phosphorus bond length in 4 increased from 2.2551(9) Å in 3 to 2.2790(13) Å in agreement with the weaker Si–P bond suggested by the coupling constants. The silylene–iron bond is slightly shortened from 2.2412(7) Å in 3 to 2.2307(10) Å in 4. The P1–W1 interatomic distance of 2.5194(8) Å is in between those of [PH3{W(CO)5}] and [PPh(SiMe3)2{W(CO)5}] (2.493(2) Å and 2.5894(5) Å, respectively).16
 | ||
| Fig. 2 Molecular structure of compound 4. Thermal ellipsoids are drawn at the 40% probability level. Hydrogen atoms except for H1 and H2, the disorder of the tBu groups and one molecule of pentane are omitted for clarity. Selected bond lengths (Å) and angles (°) in 4: Si1–P1 2.2790(13), Si1–Fe1 2.2307(10), P1–W1 2.5194(8), P1–Si1–Fe1 116.45(5), Si1–P1–W1 135.28(5). | ||
We utilized iron carbonyl as another source for a bimetallic complex to show that the coordination of phosphorus in 3 is also applicable for other than the tungsten carbonyl moiety. The homobimetallic diironcomplex [LSi{Fe(CO)4}PH2{Fe(CO)4}] (5) was obtained from 3 and [Fe2(CO)9] in a fair yield of 52% (Scheme 2). The phosphorus–hydrogen coupling constant in 5 is even a bit larger than that of the tungsten analogue 4 (1JP–H = 321 Hz and 1JP–H = 302 Hz, respectively). The 31P NMR signal of 5 is significantly downfield shifted from 4 (δ = −102.3 ppm and δ = −165.2 ppm, respectively). The 29Si{1H} NMR signal of 5 shows little difference from the doublet observed for 4 (δ = 99.2 ppm, 1JSi–P = 25 Hz and δ = 98.1 ppm, 1JSi–P = 24 Hz, respectively). The carbonyl carbons of 5 appear in the 13C{1H} NMR spectrum as a doublet at δ = 214.4 ppm (2JP–C = 17 Hz) and a singlet at δ = 214.9 ppm.
The IR spectrum of 5 exhibits two strong bands at 2055 and 2035 cm−1 with many other overlapping bands at 1970, 1955, 1938, 1924 and 1907 cm−1. The IR spectrum of 4 displayed similarly two sharp bands at 2074 and 2040 cm−1 in addition to an ill-defined broad region for the other carbonyl bands (2010–1830 cm−1). Based on the IR bands of the parent compounds, [PH3{W(CO)5}] and [PH3{Fe(CO)4}] (νCO = 2083, 1984, 1953, 1921 cm−1 and νCO = 2066, 1994, 1962 cm−1, respectively),17 the first bands of 4 and 5 at 2074 and 2055 cm−1 can be assigned to the phosphine metal carbonyl groups. Furthermore, the bands representing the silylene iron carbonyl groups are expected to be rather close to each other and 3 (νCO = 2025 cm−1). This matches well with the second bands of 4 and 5 (νCO = 2040 cm−1 and νCO = 2035 cm−1, respectively).
The PH2 moiety in 3 could also open up other pathways for the synthesis of heterobimetallic complexes. Compound 6 containing the transition metals iron and platinum, which are largely utilized in catalysis,18 can be synthesized by the insertion of platinum into the phosphorus–hydrogen bond in 60% yield (Scheme 2).19 The 1H NMR spectrum of 6 attests to this reactivity displaying the PtH signal at δ = −4.47 ppm. This doublet of doublet of doublets results from the coupling of the Pt–H with the three different phosphorus moieties (2JH–P3 = 176 Hz, 2JH–P1 = 28 Hz, 2JH–P2 = 21 Hz) and displays platinum satellites (1JH–Pt = 906 Hz). The signal of PH (P1) in 6 appears close to 5 (δ = −102.3 ppm) at δ = −105.6 ppm. This doublet of doublets in the 31P{1H} NMR spectrum exhibits the expected large and small coupling constants for trans and cis orientation (2JP1–P2 = 139 Hz and 2JP1–P3 = 11 Hz, respectively). This signal is split by the couplings to both the P–H and the Pt–H (1JP1–H1 = 214 Hz and 2JP1–H2 = 28 Hz) to a dddd in the 31P NMR spectrum. The 31P–195Pt coupling constant for P1 is significantly smaller than those of P2 and P3 (1JPt–P1 = 712 Hz, 1JP2–Pt = 2536 Hz and 1JP3–Pt = 2081 Hz). The 29Si{1H} NMR signal of 6 (δ = 114.8 ppm) is shaped as a doublet of multiplets presumably due to couplings with the triphenylphosphines. The 1JSi–P1 coupling constant in 6 (97 Hz) is higher than that of 3 (1JSi–P = 62 Hz). The 195Pt{1H} NMR spectrum of 6 exhibits the expected doublet of doublet of doublets at δ = −5035.5 ppm. The carbonyl bands in the IR spectrum of 6 are shifted to lower wave numbers compared with 3 suggesting a slightly weaker coordination of the silylene (νCO = 2014, 1933, 1890 cm−1 and νCO = 2025, 1946, 1913 cm−1 respectively). It should be noted that cyclic voltammetry studies of compounds 3 and 6 were also carried out with the results being described in the ESI.†
The iron and platinum centers in 6 are pointing in nearly opposite directions (torsion angle Fe1–Si1–P1–Pt1: 157.10(4)°) with an iron–platinum interatomic distance of 5.8864(6) Å (Fig. 3). The sum of the bond angles around platinum is close to 360°. However, the square coordination sphere of platinum is clearly distorted presumably due to steric reasons (P2–Pt1–P1 164.20(3)°, P2–Pt1–P3 102.05(3)°). The silicon–phosphorus bond in 6 is shorter than in 3, while its silicon–iron interatomic distance is larger (Si1–P1 2.2176(11) Å, Fe1–Si1 2.2917(9) Å and Si1–P1 2.2551(9) Å, Fe1–Si1 2.2412(7) Å, respectively). This supports the stronger silicon–phosphorus interaction suggested by 29Si{1H} NMR spectroscopy and the weaker coordination of the silylene found by IR spectroscopy.
 | ||
| Fig. 3 Molecular structure of compound 6. Thermal ellipsoids are drawn at the 40% probability level. Hydrogen atoms except for H1 and H2 and two molecules of thf are omitted for clarity. Selected bond lengths (Å) and angles (°) in 6: Si1–P1 2.2176(11), Fe1–Si1 2.2917(9), Pt1–P1 2.3341(7), Pt1–P2 2.2854(8), Pt1–P3 2.3301(7), P2–Pt1–P1 164.20(3), P3–Pt1–P1 92.37(3), P2–Pt1–P3 102.05(3), P1–Si1–Fe1 118.76(4), Si1–P1–Pt1 113.70(4). | ||
In summary, we succeeded in synthesizing novel heterobimetallic complexes using the phosphinosilylene ligand. Following the coordination of the silylene to iron carbonyl in 2, the trimethylsilyl groups were replaced with hydrogen to give the corresponding less bulky derivative 3, an excellent precursor for heterobimetallic complexes. Due to the increased coordination space in 3, the lone pair on phosphorus can coordinate to tungsten and iron, which produced the iron–tungsten heterobimetallic complex 4 and the homobimetallic diiron complex 5, respectively. In addition, the iron–platinum heterobimetallic complex 6 was formed via oxidative addition of platinum to the P–H bond.
The authors are exceptionally grateful to the Alexander von Humboldt foundation (Sofja Kovalevskaja Program) and the WACKER Chemie AG for financial support.
Notes and references
- (a) S. Liu, A. Motta, A. R. Mouat, M. Delferro and T. J. Marks, J. Am. Chem. Soc., 2014, 136, 10460 CrossRef CAS PubMed; (b) S. Sabater, J. A. Mata and E. Peris, Nat. Commun., 2013, 4, 2553 Search PubMed; (c) T. J. Mazzacano and N. P. Mankad, J. Am. Chem. Soc., 2013, 135, 17258 CrossRef CAS PubMed; (d) S. Liu, A. Motta, M. Delferro and T. J. Marks, J. Am. Chem. Soc., 2013, 135, 8830 CrossRef CAS PubMed; (e) N. Yamagiwa, H. Qin, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2005, 127, 13419 CrossRef CAS PubMed; (f) J. A. Mata, F. E. Hahn and E. Peris, Chem. Sci., 2014, 5, 1723 RSC.
- (a) A. Igau, H. Grutzmacher, A. Baceiredo and G. Bertrand, J. Am. Chem. Soc., 1988, 110, 6463 CrossRef CAS; (b) A. Igau, A. Baceiredo, G. Trinquier and G. Bertrand, Angew. Chem., Int. Ed. Engl., 1989, 28, 621 CrossRef; (c) A. J. Arduengo III, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361 CrossRef.
- (a) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorious, Nature, 2014, 510, 485 CrossRef CAS PubMed; (b) D. J. Nelson and S. P. Nolan, Chem. Soc. Rev., 2013, 42, 6723 RSC; (c) D. Martin, M. Melaimi, M. Soleilhavoup and G. Bertrand, Organometallics, 2011, 30, 5304 CrossRef CAS PubMed and references therein.
- (a) A. J. Arduengo III, D. Tapu and W. J. Marshall, J. Am. Chem. Soc., 2005, 127, 16400 CrossRef PubMed; (b) M. Raynal, C. S. J. Cazin, C. Vallée, H. Olivier-Bourbigou and P. Braunstein, Dalton Trans., 2009, 3824 RSC; (c) S. Sabater, J. A. Mata and E. Peris, Organometallics, 2012, 31, 6450 CrossRef CAS; (d) M. T. Zamora, M. J. Ferguson and M. Cowie, Organometallics, 2012, 31, 5384 CrossRef CAS; (e) R. Maity, H. Koppetz, A. Hepp and F. E. Hahn, J. Am. Chem. Soc., 2013, 135, 4966 CrossRef CAS PubMed; (f) D. Mendoza-Espinosa, B. Donnadieu and G. Bertrand, Chem. – Asian J., 2011, 6, 1099 CrossRef CAS PubMed; (g) J. Ruiz and A. F. Mesa, Chem. – Eur. J., 2012, 18, 4485 CrossRef CAS PubMed.
- G. Tan, S. Enthaler, S. Inoue, B. Blom and M. Driess, Angew. Chem., Int. Ed., 2015, 54, 2214 CrossRef CAS PubMed.
- (a) L. Álvarez-Rodríguez, J. A. Cabeza, P. García-Álvarez and D. Polo, Coord. Chem. Rev., 2015, 300, 1 CrossRef; (b) B. Blom, M. Stoelzel and M. Driess, Chem. – Eur. J., 2013, 19, 40 CrossRef CAS PubMed; (c) R. S. Ghadwal, R. Azhakar and H. W. Roesky, Acc. Chem. Res., 2013, 46, 444 CrossRef CAS PubMed; (d) H. Ogino, Chem. Rec., 2002, 2, 291 CrossRef CAS PubMed and references therein.
- B. Blom, D. Gallego and M. Driess, Inorg. Chem. Front., 2014, 1, 134 RSC.
- (a) W. Wang, S. Inoue, S. Yao and M. Driess, J. Am. Chem. Soc., 2010, 132, 15890 CrossRef CAS PubMed; (b) W. Wang, S. Inoue, S. Enthaler and M. Driess, Angew. Chem., Int. Ed., 2012, 51, 6167 CrossRef CAS PubMed; (c) N. C. Breit, T. Szilvási, T. Suzuki, D. Gallego and S. Inoue, J. Am. Chem. Soc., 2013, 135, 17958 CrossRef CAS PubMed; (d) W. Wang, S. Inoue, E. Irran and M. Driess, Angew. Chem., Int. Ed., 2012, 51, 3691 CrossRef CAS PubMed; (e) A. Brück, D. Gallego, W. Wang, E. Irran, M. Driess and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 11478 CrossRef PubMed; (f) D. Gallegeo, A. Brück, E. Irran, F. Meier, M. Kaupp, M. Driess and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 15617 CrossRef PubMed; (g) C. I. Someya, M. Haberberger, W. Wang, S. Enthaler and S. Inoue, Chem. Lett., 2013, 42, 286 CrossRef CAS; (h) D. Gallego, S. Inoue, B. Blom and M. Driess, Organometallics, 2014, 33, 6885 CrossRef CAS.
- (a) H. Sakaba, H. Oike, M. Kawai, M. Takami, C. Kabuto, M. Ray, Y. Nakao, H. Sato and S. Sakaki, Organometallics, 2011, 30, 4515 CrossRef CAS; (b) G. Tan, B. Blom, D. Gallego and M. Driess, Organometallics, 2014, 33, 363 CrossRef CAS.
- (a) S. D. Grumbine, T. D. Tilley and A. L. Rheingold, J. Am. Chem. Soc., 1993, 115, 358 CrossRef CAS; (b) U. Bodensieck, P. Braunstein, W. Deck, T. Faure, M. Knorr and C. Stern, Angew. Chem., Int. Ed. Engl., 1994, 33, 2440 CrossRef; (c) K. H. Pannell, H. K. Sharma, R. N. Kapoor and F. Cervantes-Lee, J. Am. Chem. Soc., 1997, 119, 9315 CrossRef CAS.
- (a) H. H. Karsch, U. Keller, S. Gamper and G. Müller, Angew. Chem., Int. Ed. Engl., 1990, 29, 295 CrossRef; (b) C.-W. So, H. W. Roesky, P. M. Gurubasavaraj, R. B. Oswald, M. T. Gamer, P. G. Jones and S. Blaurock, J. Am. Chem. Soc., 2007, 129, 12049 CrossRef CAS PubMed; (c) S. Inoue, W. Wang, C. Präsang, M. Asay, E. Irran and M. Driess, J. Am. Chem. Soc., 2011, 133, 2868 CrossRef CAS PubMed; (d) R. Azhakar, R. S. Ghadwal, H. W. Roesky, H. Wolf and D. Stalke, Organometallics, 2012, 31, 4588 CrossRef CAS; (e) K. Hansen, T. Szilvási, B. Blom, E. Irran and M. Driess, Chem. – Eur. J., 2015, 21, 18930 CrossRef CAS PubMed; (f) H. Cui, J. Zhang, Y. Tao and C. Cui, Inorg. Chem., 2016, 55, 46 CrossRef CAS PubMed.
- (a) R. Azhakar, K. Pröpper, B. Dittrich and H. W. Roesky, Organometallics, 2012, 31, 7586 CrossRef CAS; (b) N. C. Breit, T. Szilvási and S. Inoue, Chem. – Eur. J., 2014, 20, 9312 CrossRef CAS PubMed; (c) N. C. Breit, T. Szilvási and S. Inoue, Chem. Commun., 2015, 52, 11272 RSC.
- (a) K. Junold, J. A. Baus, C. Burschka, T. Vent-Schmidt, S. Riedel and R. Tacke, Inorg. Chem., 2013, 52, 11593 CrossRef CAS PubMed; (b) F. M. Mück, D. Kloß, J. A. Baus, C. Burschka and R. Tacke, Chem. – Eur. J., 2014, 20, 9620 CrossRef PubMed.
- W. Yang, H. Fu, H. Wang, M. Chen, Y. Ding, H. W. Roesky and A. Jana, Inorg. Chem., 2009, 48, 5058 CrossRef CAS PubMed.
- D. M. Schubert and A. D. Norman, Inorg. Chem., 1985, 24, 1107 CrossRef CAS.
- (a) G. Frenking, K. Wichmann, N. Fröhlich, J. Grobe, W. Golla, D. Le Van, B. Krebs and M. Läge, Organometallics, 2002, 21, 2921 CrossRef CAS; (b) C. P. Rooney, J. L. Wade, A. C. Hinkle, R. M. Stolley, S. M. Miller and M. L. Helm, Main Group Chem., 2008, 7, 155 CrossRef CAS.
- E. O. Fischer, E. Louis, W. Bathelt and J. Müller, Chem. Ber., 1969, 102, 2547 CrossRef CAS.
- (a) I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170 CrossRef CAS PubMed; (b) A. Fürstner, Chem. Soc. Rev., 2009, 38, 3208 RSC.
- (a) U. Vogel and M. Scheer, Z. Anorg. Allg. Chem., 2001, 627, 1593 CrossRef CAS; (b) C. A. Jaska, H. Dorn, A. J. Lough and I. Manners, Chem. – Eur. J., 2003, 9, 271 CrossRef CAS PubMed; (c) U. Vogel, K.-C. Schwan, P. Hoemensch and M. Scheer, Eur. J. Inorg. Chem., 2005, 1453 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental procedures and characterization methods, NMR spectra, mass spectra, IR spectra, cyclic voltammograms, and crystallographic data. CCDC 1435730 (2), 1435732 (3), 1435731 (4), and 1436733 (6). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc00601a |
| This journal is © The Royal Society of Chemistry 2016 |