
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Full chirality transfer in the synthesis of hindered tertiary boronic esters under in situ lithiation–borylation conditions†
D. J.
Blair‡
,
S.
Zhong‡
,
M. J.
Hesse
,
N.
Zabaleta
,
E. L.
Myers
and
V. K.
Aggarwal
*
School of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS, UK. E-mail: v.aggarwal@bristol.ac.uk; Fax: +44 (0)117 929 8611; Tel: +44 (0)117 954 6315
First published on 22nd March 2016
Abstract
Hindered tertiary neopentyl glycol boronic esters can be prepared by using in situ lithiation–borylation of enantiopure secondary benzylic carbamates at −20 °C with full chirality transfer.
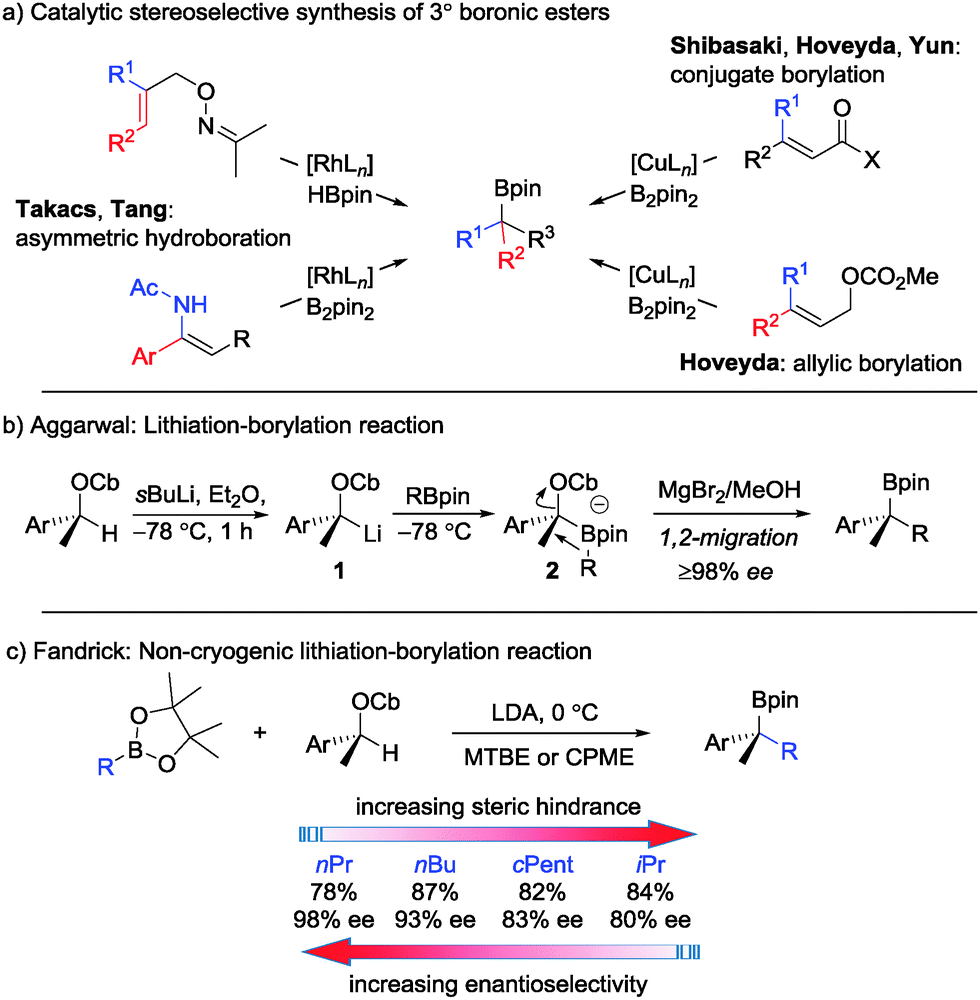
Boronic esters are versatile intermediates in synthesis and there are now numerous methods for their preparation in enantioenriched form.1 In the case of tertiary boronic esters, which are more difficult to prepare, a number of stereoselective and stereospecific methods have emerged over recent years (Scheme 1a (ref. 2) and Scheme 1b (ref. 3)). The stereospecific lithiation–borylation of enantiopure secondary carbamates (Scheme 1b), which has been developed in our research group,3 has been employed by others4 and indeed has even been scaled up to 24 kg.5 For this scale-up, the cryogenic conditions commonly employed (sBuLi, −78 °C)6 presented challenges. However, Fandrick5 discovered that the carbamate could be deprotonated by a weaker base (LDA) and that this deprotonation could be conducted in the presence of pinacol boronic esters (in situ conditions) at elevated temperatures (0 °C), to give the corresponding tertiary boronic esters with high levels of enantiospecificity (Scheme 1c). At such an elevated temperature, having the boronic ester present in the reaction mixture during the deprotonation prevents epimerisation and/or decomposition of lithiated carbamate 1.
 | ||
| Scheme 1 State of the art: synthesis of tertiary boronic esters. | ||
With more hindered secondary boronic esters, such as iPrBpin, lower levels of enantiospecificity were observed, presumably due to reversible formation of boronate complex 2. Such a process would return the sensitive lithiated carbamate 1, which would undergo racemisation and recombination with the boronic ester, thus leading to reduced stereoselectivity ∼80% es (Scheme 1b). We have previously found that the addition of MgBr2/methanol following boronate complex formation enhances the rate of 1,2-migration and quenches any lithiated carbamate generated by the reverse process thereby leading to high yields and high stereoselectivity.3a Unfortunately, the addition of MgBr2 in methanol is not compatible with an in situ lithiation–borylation reaction. Herein, we address the issue of low selectivity with hindered boronic esters and show that by using neopentyl glycol boronic esters7 and LTMP (lithium 2,2,6,6-tetramethylpiperidine) as a base, high levels of enantiospecificity can now be achieved even with some of the most hindered boronic esters under non-cryogenic conditions.
During our investigations of lithiation–borylation methodology we found that the nature of the ligand on boron sometimes affected the enantioselectivity of the process.3b,8 This is most dramatically illustrated in the case of the propargylic carbamate 3 where upon moving from the pinacol to the ethylene glycol based isopropyl boronic ester, enantiospecificity increased from 4% to 100% (Scheme 2).8a Presumably, as the steric hindrance around boron was reduced, the boronate complex became less prone to reversibility and consequently the intermediate lithiated carbamate suffered less racemisation.
 | ||
| Scheme 2 Influence of the boron ligand on the stereochemical outcome of the lithiation–borylation of 3; these results are taken from ref. 8a and are shown for comparison to results below (see Scheme 7). | ||
We therefore explored Fandrick's in situ conditions5 with iPrBneo in place of iPrBpin. These conditions gave tertiary alcohol 5 from 4 with full stereospecificity (100% es, Scheme 3), a substantial improvement on that obtained using the pinacol boronate (80% es).5 Other bases were tested and LTMP led to a higher yield (74%). Further improvement in the yield was realised by reducing the temperature from 0 °C to −20 °C, resulting in 5 being isolated in 90% yield and 100% es.
 | ||
| Scheme 3 Conditions for the homologation of neopentyl glycol boronic esters. | ||
Our optimised conditions were applied to a range of otherwise challenging carbamates (Scheme 4). As noted by Fandrick, the in situ conditions involving an amide base in place of an organolithium base enables aryl bromides and iodides to be employed, and so these were initially tested. Using our conditions, these substrates gave the corresponding tertiary alcohols 6 and 7 in high yields and complete enantiospecificity. The para-phenyl-substituted carbamate is especially prone to racemisation and using iPrBpin gave 8 in low enantiospecificity (83% es). However, using the neopentyl glycol boronic ester, 8 was again obtained in high yield and enantiospecificity. Electron-withdrawing groups on the aromatic ring engenders reversibility in the formation of the boronate complex, thus rendering the lithiated carbamate more prone to racemisation. We found that although a single meta-CF3 group was tolerated, enabling formation of 9 with excellent enantiospecificity, two meta-CF3 groups was a step too far and led to essentially racemic product (10).9 A hindered ester was compatible with our conditions and gave tertiary alcohol 11 with complete enantiospecificity. This functional group would not have been compatible with the preformed lithiated carbamate. Unfortunately, the use of ortho-substituted benzylic carbamates did not lead to the expected products. In contrast, we have previously shown these carbamates do give the expected products in good yield and near-complete enantiospecificity when subjected to our cryogenic lithiation–borylation conditions.3a,10
 | ||
| Scheme 4 Carbamate scope for the in situ lithiation–borylation of iPrBneo. | ||
Because the stereoselectivity of the in situ lithiation–borylation reaction is affected by the steric bulk of the boronic ester substituent (R group), we tested a range of boronic esters of varying steric demand and compared both the pinacol (A) and neopentyl glycol (B) derivatives (Scheme 5). For unhindered nBu (13) and cyclopropyl (14) boronic esters high enantiospecificity was observed by using pinacol boronic esters (98% es), with neopentyl glycol boronic esters behaving similarly. Surprisingly, with unhindered allyl boronic esters (15) the pinacol derivative gave low es (82%) whilst the neopentyl glycol ester provided essentially complete enantiospecificity.
 | ||
| Scheme 5 Influence of boronic ester substituent on the in situ lithiation–borylation reaction of 12. a Because the use of the pinacol boronic ester gave tertiary alcohol 14 with complete diastereospecificity, the corresponding experiment with the neopentyl glycol boronic ester was not carried out. | ||
To explore the limits in steric bulk that could be tolerated we turned to 3-pentyl boronic esters.11 Reaction of 3-pentyl-Bpin with 12 under our in situ conditions gave only traces of 16 with poor enantioselectivity. Simply switching to the corresponding neopentyl glycol ester significantly increased both the yield and selectivity, thus highlighting the advantages associated with neopentyl glycol derivatives, particularly in their application to hindered systems.
We have previously shown that secondary benzylic pinacol boronic esters form reversible boronate complexes with secondary benzylic carbamates leading to loss of both diastereo- and enantioselectivity.8c The application of in situ conditions to the reaction of (R)-1-phenylethyl pinacol boronic ester with 12 gave 17 in high yield (90%, Scheme 6), high enantiospecificity (100% es) with respect to the boronic ester starting material, but low diastereoselectivity (85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 dr).12 Simply switching to the corresponding neopentyl glycol boronic ester gave 17 in 94:6 dr, 100% es and high yield. In contrast, reaction of 12 with both pinacol and neopentyl glycol (S)-1-phenylethyl boronic esters gave 18 in high yield and selectivity (≥95:5 dr, 100% es).13 Evidently, there is a significant matched/mis-matched effect operating under the reversible conditions with the pinacol boronic esters that can be minimised by using the neopentyl glycol boronic esters.
:15 dr).12 Simply switching to the corresponding neopentyl glycol boronic ester gave 17 in 94:6 dr, 100% es and high yield. In contrast, reaction of 12 with both pinacol and neopentyl glycol (S)-1-phenylethyl boronic esters gave 18 in high yield and selectivity (≥95:5 dr, 100% es).13 Evidently, there is a significant matched/mis-matched effect operating under the reversible conditions with the pinacol boronic esters that can be minimised by using the neopentyl glycol boronic esters.
 | ||
| Scheme 6 The influence of boronic ester diol on the stereochemical outcome of homologation of (R)- and (S)-1-phenylethyl boronic esters with 12 under in situ conditions using pinacol (A) and neopentyl glycol (B) boronic esters. | ||
As noted above, for substrates that are especially prone to reversibility in boronate formation and therefore racemisation (e.g.8), the in situ conditions using neopentyl glycol boronic esters can lead to considerably higher levels of enantiospecificity. We therefore tested our in situ conditions with the secondary propargylic carbamate 19, a substrate that only gave 81% es under conditions where the lithiated carbamate was preformed8a (Scheme 7). Under the new in situ conditions the tertiary propargylic alcohol 20 was obtained in high yield and excellent enantiospecificity (98% es).14 This highlights the broad applicability of the new in situ lithiation–borylation protocol.15
 | ||
| Scheme 7 Enhanced stereospecificity using in situ conditions in the lithiation–borylation reaction of secondary propargylic carbamates. | ||
In summary, we have found that almost complete enantiospecificity can be achieved in the lithiation–borylation reactions of secondary benzylic carbamates under in situ conditions when neopentyl glycol boronic esters are used in place of pinacol boronic esters. These conditions expand the range of tertiary boronic esters that can be prepared with very high selectivity with both increased functional-group and steric tolerance. The improved stereoselectivity results from reduced reversibility in boronate complex formation, a process that otherwise causes racemisation of the sensitive lithiated carbamate.
We thank European Research Council (FP7/2007-2013, ERC grant no. 246785; H2020/2015-2020, ERC grant no. 670668), and EPSRC (EP/I038071/1) for financial support. N. Z. thanks UPV/EHU predoctoral mobility program. S. Z. thanks the EPSRC-funded Bristol Synthesis Centre for Doctoral Training (EP/L015366/1).
Notes and references
- (a) Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials (Volume 1 and 2), ed. D. G. Hall, Wiley-VCH Verlag GmbH & Co. KGaA, 2nd edn, 2011 Search PubMed; (b) Synthesis and Application of Organoboron Compounds, ed. E. Fernandez and A. Whiting, Springer International Publishing, 2015 Search PubMed.
- (a) I. H. Chen, L. Yin, W. Itano, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 6746 Search PubMed; (b) J. M. O'Brien, K. Lee and A. H. Hoveyda, J. Am. Chem. Soc., 2010, 132, 10630 CrossRef PubMed; (c) X. Feng and J. Yun, Chem. – Eur. J., 2010, 16, 13609 CrossRef CAS PubMed; (d) S. Radomkit and A. H. Hoveyda, Angew. Chem., Int. Ed., 2014, 126, 3455 CrossRef; (e) N. Hu, G. Zhao, Y. Zhang, X. Liu, G. Li and W. Tang, J. Am. Chem. Soc., 2015, 137, 6746 CrossRef CAS PubMed ; For recent syntheses of racemic tertiary boronic esters see:; (f) A. J. Wommack and J. S. Kingsbury, Tetrahedron Lett., 2014, 55, 3163 CrossRef CAS; (g) K. Hong, X. Liu and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 10581 CrossRef CAS PubMed.
- (a) J. L. Stymiest, V. Bagutski, R. M. French and V. K. Aggarwal, Nature, 2008, 456, 778 CrossRef CAS PubMed; (b) V. Bagutski, R. M. French and V. K. Aggarwal, Angew. Chem., Int. Ed., 2010, 49, 5142 CrossRef CAS PubMed ; For reviews see: ; (c) S. P. Thomas, R. M. French, V. Jheengut and V. K. Aggarwal, Chem. Rec., 2009, 9, 24 CrossRef CAS PubMed; (d) D. Leonori and V. K. Aggarwal, Acc. Chem. Res., 2014, 47, 3174 CrossRef CAS PubMed.
- (a) B. W. Glasspoole, K. Ghozati, J. W. Moir and C. M. Crudden, Chem. Commun., 2012, 48, 1230 RSC; (b) R. D. Grigg, J. W. Rigoli, R. Van Hoveln, S. Neale and J. M. Schomaker, Chem. – Eur. J., 2012, 18, 9391 CrossRef CAS PubMed; (c) P. A. Evans, S. Oliver and J. Chae, J. Am. Chem. Soc., 2012, 134, 19314 CrossRef CAS PubMed; (d) P. A. Evans and S. Oliver, Org. Lett., 2013, 15, 5626 CrossRef CAS PubMed; (e) Y. Li, S. Chakrabarty and A. Studer, Angew. Chem., Int. Ed., 2015, 54, 3587 CrossRef CAS PubMed; (f) B. W. H. Turnbull, S. Oliver and P. A. Evans, J. Am. Chem. Soc., 2015, 137, 15374 CrossRef CAS PubMed.
- (a) K. R. Fandrick, J. A. Mulder, N. D. Patel, J. Gao, M. Konrad, E. Archer, F. G. Buono, A. Duran, R. Schmid, J. Daeubler, J.-N. Desrosier, X. Zeng, S. Rodriguez, S. Ma, B. Qu, Z. Li, D. R. Fandrick, N. Grinberg, H. Lee, T. Dosanac, H. Takahashi, Z. Chen, A. Bartolozzi, P. Nemoto, C. A. Busacca, J. J. Song, N. K. Yee, P. E. Mahaney and C. H. Senanayake, J. Org. Chem., 2014, 80, 1651 CrossRef PubMed; (b) K. R. Fandrick, N. D. Patel, J. A. Mulder, J. Gao, M. Konrad, E. Archer, F. G. Buono, A. Duran, R. Schmid, J. Daeubler, D. R. Fandrick, S. Ma, N. Grinber, H. Lee, C. A. Busacca, J. J. Song, N. K. Yee and C. H. Senanayake, Org. Lett., 2014, 16, 4360 CrossRef CAS PubMed; (c) K. R. Fandrick, J. J. Gao, J. A. Mulder, N. D. Patel and X. Zeng, WO 2013119751, 2013 Search PubMed.
- (a) D. Hoppe, A. Carstens and T. Kramer, Angew. Chem., Int. Ed. Engl., 1990, 29, 1424 CrossRef; (b) A. Carstens and D. Hoppe, Tetrahedron, 1994, 50, 6097 CrossRef CAS.
- Neopentyl glycol boronates, which are less sterically hindered than the corresponding pinacol boronic esters, have found application in other organoboron transformations besides lithiation–borylation. For selected examples, see: (a) F. J. Lawlor, N. C. Norman, N. L. Pickett, E. G. Robins, P. Nguyen, G. Lesley, T. B. Marder, J. A. Ashmore and J. C. Green, Inorg. Chem., 1998, 37, 5282 CrossRef CAS; (b) S. Ueno, N. Chatani and F. Kakiuchi, J. Am. Chem. Soc., 2007, 129, 6098 CrossRef CAS PubMed; (c) K. Huang, D.-G. Yu, S.-F. Zheng, Z.-H. Wu and Z.-J. Shi, Chem. – Eur. J., 2011, 17, 786 CrossRef CAS PubMed; (d) N. Zhang, D. J. Hoffman, N. Gutsche, J. Gupta and V. Percec, J. Org. Chem., 2012, 77, 595 Search PubMed; (e) M. Tobisu, H. Kinuta, Y. Kita, E. Rémond and N. Chatani, J. Am. Chem. Soc., 2012, 134, 115 CrossRef CAS PubMed; (f) S. C. Matthew, B. W. Glasspoole, P. Eisenberger and C. M. Crudden, J. Am. Chem. Soc., 2014, 136, 5828 CrossRef CAS PubMed; (g) X.-W. Liu, J. Echavarren, C. Zarate and R. Martin, J. Am. Chem. Soc., 2015, 137, 12470 CrossRef CAS PubMed; (h) K. Feeney, G. Berionni, H. Mayr and V. K. Aggarwal, Org. Lett., 2015, 17, 2614 CrossRef CAS PubMed; (i) C. Zarate, R. Manzano and R. Martin, J. Am. Chem. Soc., 2015, 137, 6754 CrossRef CAS PubMed; (j) L. Fang, L. Yan, F. Haeffner and J. P. Morken, J. Am. Chem. Soc., 2016, 138, 2508 CrossRef CAS PubMed.
- (a) B. M. Partridge, L. Chausset-Boissarie, M. Burns, A. P. Pulis and V. K. Aggarwal, Angew. Chem., Int. Ed., 2012, 51, 11795 CrossRef CAS PubMed; (b) A. P. Pulis, D. J. Blair, E. Torres and V. K. Aggarwal, J. Am. Chem. Soc., 2013, 135, 16054 CrossRef CAS PubMed; (c) S. Roesner, D. J. Blair and V. K. Aggarwal, Chem. Sci., 2015, 6, 3718 RSC.
- Reducing the steric bulk of the boronic ester reaction partner from iPr to Et had no effect on the stereochemical outcome.
- The ortho-bromo isomer of 6 and an ortho-methyl substrate (not depicted), did not lead to the expected products. The former substrate did not react whereas the latter gave a mixture of isomeric species derived from its fragmentation into the corresponding ortho-quinodimethane followed by (4+2) cycloaddition. See the ESI,† for further details on this side reaction.
- tBu boronic esters were also tested but both pinacol and neopentyl glycol esters failed to react under the in situ conditions.
- Enantiospecificity was determined for the major diastereoisomer.
- No epimerisation of the secondary boronic ester stereocentre was observed in either 17 or 18 with pinacol or neopentyl glycol esters. The presence of an electron-withdrawing group in 12 must bias the corresponding boronate complex towards fragmentation to Li-12 rather than fragmentation of the C–B bond of the secondary boronic ester substrate to form a benzylic anion.
- For the propargylic carbamates, unfortunately the use of a trialkylsilyl substituent in place of a t-butyl group led to desilylated products not incorporating the boronic ester organic group.
- We have previously shown that when substituted allylic carbamates are subjected to lithiation–borylation reactions under cryogenic conditions with neopentyl glycol boronic esters as partners, a mixture of products derived from α- and γ-borylation are obtained: (a) A. P. Pulis, PhD thesis, University of Bristol, 2012. However, the use of pinacol boronic esters lead to predominantly the required α-borylation: ; (b) A. P. Pulis and V. K. Aggarwal, J. Am. Chem. Soc., 2012, 134, 7570 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental details and characterisation. See DOI: 10.1039/c6cc00536e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |