
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Correlation between Cu ion migration behaviour and deNOx activity in Cu-SSZ-13 for the standard NH3-SCR reaction†
A. M.
Beale
*ab,
I.
Lezcano-Gonzalez
ab,
W. A.
Slawinksi
c and
D. S.
Wragg
c
aDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: andrew.beale@ucl.ac.uk
bResearch Complex at Harwell, Rutherford Appleton Laboratory, Harwell Science and Innovation Campus, Harwell, Didcot, Oxon, Ox11 0FA, UK
cINGAP Centre for Research Based Innovation, Department of Chemistry, University of Oslo, N-0315 Oslo, Norway
First published on 8th April 2016
Abstract
Here we present the results of a synchrotron-based in situ, time-resolved PXRD study during activation of two Cu-SSZ-13 catalysts under O2/He and one during standard NH3-SCR reaction conditions to obtain insight into the behaviour of Cu ions. The results obtained indicate that deNOx activity is inexorably linked with occupancy of the zeolite 6r.
Zeolite-based ammonia-selective catalytic reduction (NH3-SCR) is a widely employed technology for NOx abatement particularly from mobile sources.1,2 Early studies by Iwamoto et al.,3 focused on metal-promoted medium pore zeolites, such as MFI and BEA-type zeolites which, despite displaying a high activity at low temperatures (<300 °C), were susceptible to hydrothermal deactivation. In recent times it has been shown that small-pore zeolites such as Cu-exchanged SSZ-13 (Cu-SSZ-13) and SAPO-34 exhibit the required high activity at low temperatures but also enhanced hydrothermal stability compared to other medium-pore zeolite topologies, leading to a successful commercialisation.1,2
SSZ-13, which has the CHA topology, is a three-dimensional small pore zeolite composed of double 6-membered rings (d6r) of SiO4 tetrahedra, connected by four-membered rings in an AABBCC sequence, this creates a framework in which large cha cages are defined by the d6r and 8-membered rings (8r).4 Much research has been undertaken to determine the nature and location of Cu species within the zeolite structure. Recently there have been proposals that oxo-clusters or dimeric species may play an important role in NO oxidation (as a first step in the NOx removal process) but there is much evidence that isolated Cu ions located within the micropore volume are catalytically important for SCR.1 However, the precise role(s) of the ion-exchanged cations in the catalytic process is (are) still not clear. This issue is further complicated by the fact that Cu ions, like many metal cations inside a microporous volume, tend to migrate during heat treatment;4–6 a combination of in situ PXRD and PXRD/XAFS measurements have broadly shown that at room temperature Cu2+ ions are located in the cages/8r windows of the SSZ-13 structure as hexa-aqua species but that on heating they dehydrate, become mobile and move into the corners of the 6r.7,8 However, other studies have suggested that the location and occupancy of Cu species in these sites is affected by Cu loading.9 Furthermore, it has also been indicated that single Cu ions located in either site maybe active, but that those in the 8r in particular are easier to reduce and potentially therefore more active.1,8
Clearly then there is an urgent need to understand the principle location and the impact (if any) of Cu migration in relation to NH3-SCR activity. To this end we have performed synchrotron-based in situ, time-resolved PXRD (powder X-ray diffraction) measurements during calcination (up to 500 °C) and under standard NH3-SCR conditions (at 200 °C) in order to better understand the nature/behaviour of the Cu ions as active sites in the standard NH3-SCR reaction. We investigated two types of Cu-SSZ-13 sample labelled Cu-SSZ-13-1 and Cu-SSZ-13-2 respectively. The principal difference between these two samples is their differing NH3-SCR activity that has been brought about by differences in their post-synthetic treatment history; Cu-SSZ-13-1 has undergone a combination of a single ion exchange and calcination, whereas Cu-SSZ-13-2 underwent this two-step process four times.10 As can be seen in Fig. 1, the catalytic activity for this second sample is much greater than for the first most likely due to differences in Al3+ distribution.10,11 We now show, using in situ PXRD, how the population of Cu in the 6r at low temperatures (T < 300 °C) can be correlated with improved catalytic activity.
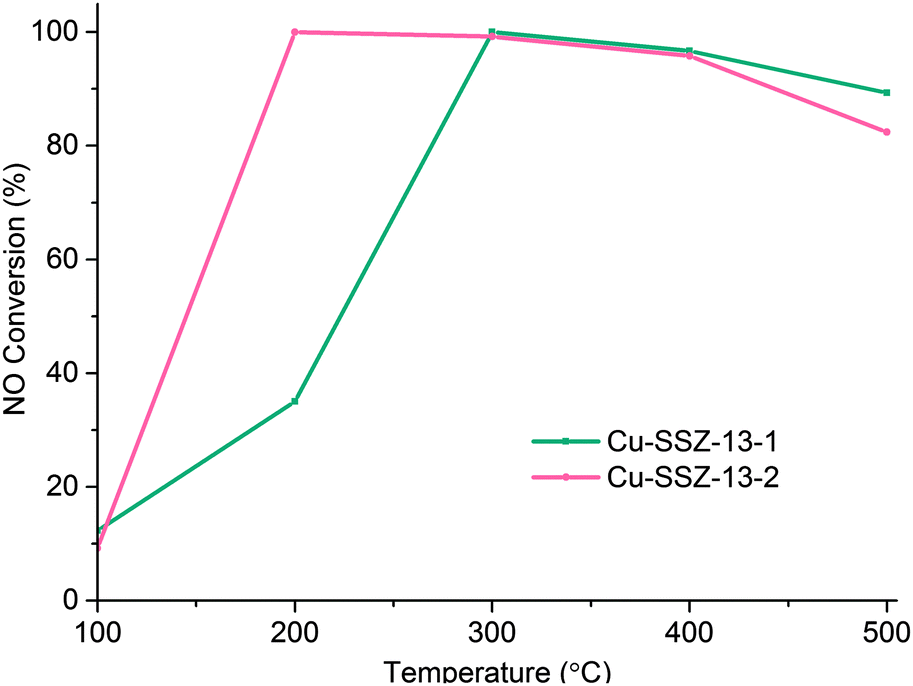 | ||
Fig. 1 NOx conversion for Cu-SSZ-13-1 and Cu-SSZ-13-2 at different temperatures, using a GHSV of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 and a feed composition of 1000 ppm NO, 1000 ppm NH3 and 5% O2. 000 h−1 and a feed composition of 1000 ppm NO, 1000 ppm NH3 and 5% O2. | ||
Fig. S2a (in the ESI†) contains a stack-plot of PXRD patterns recorded during activation of Cu-SSZ-13-1 to 500 °C whilst Fig. S2b (ESI†) shows the observed, calculated, and difference pattern obtained from the Rietveld refinement of PXRD data on Cu-SSZ-13-1 at 100 °C. Fig. S2c (ESI†) contains the results from the Rietveld refinement of Cu-SSZ-13-1 recorded at 500 °C. The reflections in all datasets can be readily indexed with the Cu-SSZ-13 unit cell.6 The major differences between the room temperature diffraction pattern and that recorded at 500 °C is an increase in intensity and a shift towards higher angles of many of the Bragg reflections at low 2θ with time/temperature, the time-resolved in situ PXRD data revealed that these changes occur throughout the calcination process. These changes have previously been observed during calcination of similar materials and have been proposed to occur as a result of dehydration of the zeolite, migration of the Cu ions within the framework and negative thermal expansion.7
In order to verify the general observations made from the stack-plot data we performed a Rietveld and difference Fourier analysis on the Cu-SSZ-13 data as a function of temperature. The corresponding experimental and refinement details (experimental parameters and goodness of fit factors) listed in Table S2 (ESI†). Tables S3 and S4 (ESI†) contain the corresponding bond distances and angles as well as the lattice parameters obtained from the Rietveld refinement. The average Si(Al)–O bond lengths of ∼1.60 Å with a O–T–O bond angle ∼109.3° are in good agreement with previously reported literature values for framework ions (Si4+/Al3+) occupying tetrahedral sites.6 Difference Fourier mapping was used to determine that Cu ions (ostensibly in the 2+ oxidation state) are located exclusively in/around the 8r of the Cu-SSZ-13 structure at room temperature (see Fig. 2a).12,13 At 500 °C, Cu ions occupy predominantly (i.e. ∼80% of the Cu) the planar environment of the 6r (see Fig. 2b).6,14 The Cu ion is coordinated to three (symmetrically equivalent) oxygens at a distance of 2.32 Å, although as has been shown previously, this environment is a symmetry-averaged position for three Cu2+ ions ensconced in the corners of the 6r with a Cu–O distance of ∼1.95 Å.14 The difference in Cu ion location at 100 and 500 °C is consistent with previous studies on this material and indicate that Cu ions migrate with increasing temperature.6,12,14 In order to see at which point the Cu occupancies begin to change we turn our attention to the time-resolved PXRD data processed using parametric methods.15 The results for the change in Cu site occupancy are shown in Fig. 3a and reveal that the changes begin on the application of heat; this change in occupancy is initially rapid due, in part (up to ∼110 °C), to the effect of dehydration of the [Cu(H2O)6]2+ complex. On reaching ∼220 °C the occupancy of the 6r finally exceed that of the 8r. Heating above 220 °C sees the 6r continue to be populated at the expense of the 8r but at a slower rate and on reaching 500 °C (end of the activation) some Cu remains in the 8r. The contraction in unit cell volume (Fig. S3, ESI†) observed when calcination reaches 500 °C is consistent with previously observed dehydration and negative thermal expansion in microporous materials possessing the CHA structure.16,17
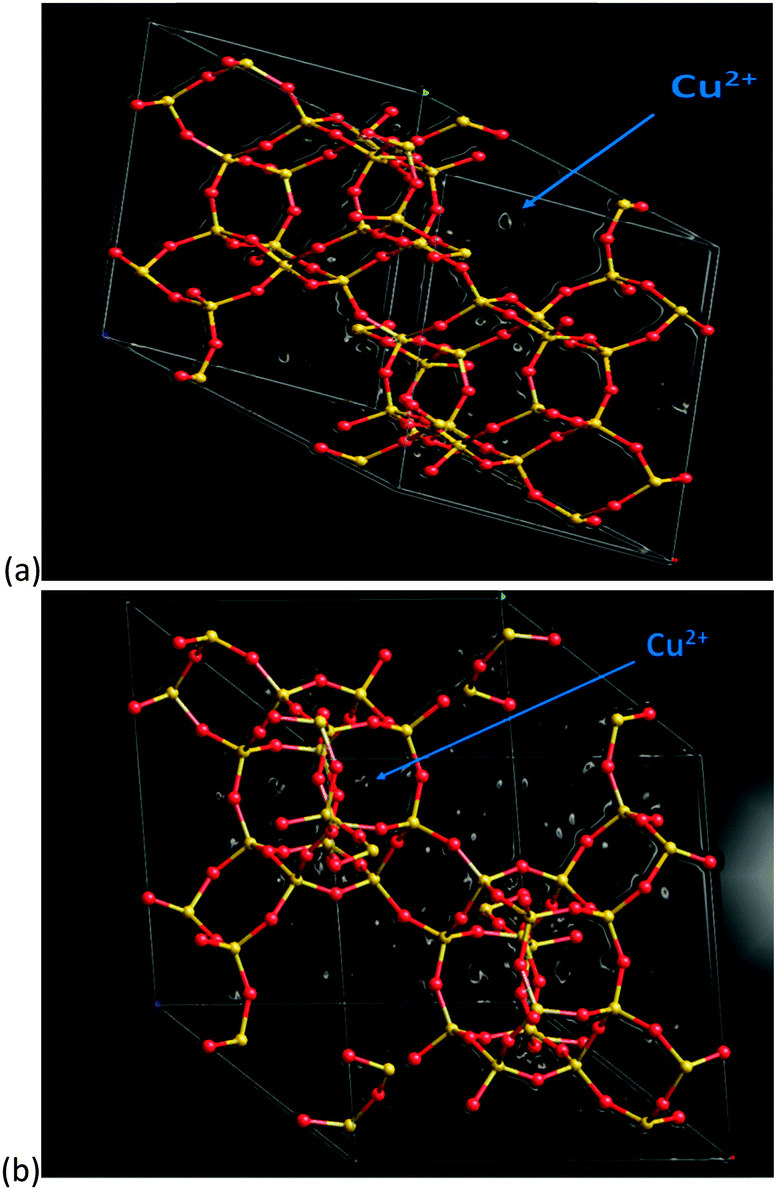 | ||
| Fig. 2 Difference Fourier map of Cu-SSZ-13-1 sample recorded at 100 °C (a) and 500 °C (b) respectively. Atom colour key (framework): Si4+/Al3+ – yellow, O2− – red. White circular objects represent regions of electron density of which the larger components are located in the 8r in (a) and in the 6r in (b) which we assign to Cu2+ ions (indicated). Note that the 100 °C structure was used rather than room temperature as the cha cages are free of adsorbed water, making data analysis easier. | ||
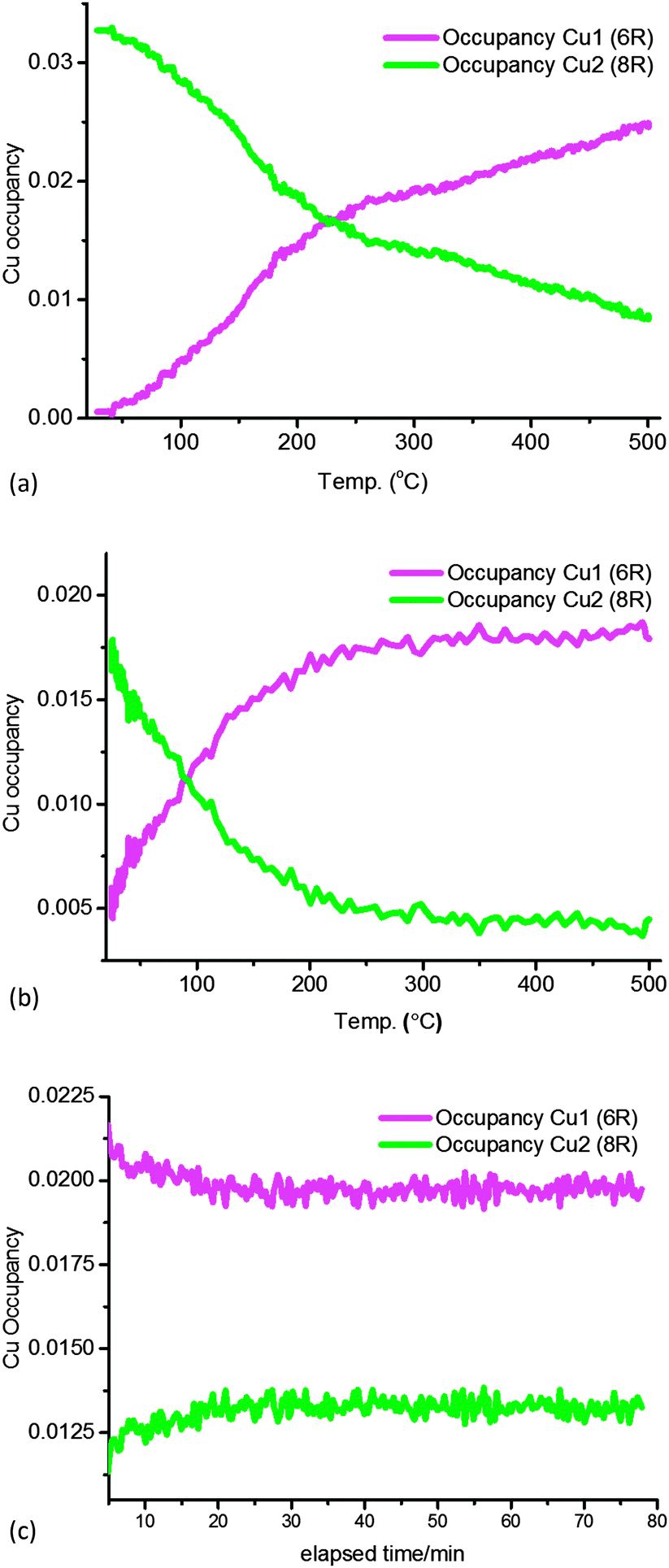 | ||
| Fig. 3 Plots of Cu site occupancy in the 8r (green) and 6r (magenta) as a function of temperature up to 500 °C in Cu-SSZ-13-1 (a) and Cu-SSZ-13-2 during (b) activation in dry air. In (c) are the data recorded for sample Cu-SSZ-13-1 during the standard NH3-SCR reaction at constant temperature (200 °C). | ||
A stack plot of the PXRD data recorded during calcination of Cu-SSZ-13-2 is shown in Fig. S4a (ESI†) with the results from the Rietveld refinement of the data recorded at 100 and 500 °C shown in Fig. S4b and c (ESI†) with details of the derived structural parameters given in Tables S5–S7 (ESI†). As with the Cu-SSZ-13-1 sample, changes are observed in reflection intensity and 2θ position associated with zeolite/Cu dehydration. Rietveld analysis of the data again revealed the presence of Cu2+ ions in the vicinity of the 8r but unlike Cu-SSZ-13-1, a significant portion (∼28%) of the total Cu content is already present in the 6r at room temperature (see Fig. 3b). Heating the sample to 500 °C sees again a depopulation of the 8r in favour of the 6r although remarkably this is essentially complete on reaching ∼300 °C with the point of crossover for the site occupancies reached at the rather low temperature of 100 °C. Note also that according to in situ XAFS data the Cu K-edge XANES and EXAFS data for both samples during activation are almost identical at 500 °C and therefore consistent with a local structure as previously reported (Fig. S7, ESI†).10,14
The standard SCR reaction proceeds as follows (eqn (1)):
| 4NH3(g) + 4NO(g) + O2(g) → 4N2(g) + 6H2O(g) | (1) |
The general behaviour observed in all three experiments pertaining to the behaviour of Cu can be described in terms of a dehydration of [Cu(H2O)6]2+ species accompanied by migration of ions from the vicinity of the 8r to the 6r. The driving force for this migration appears to be the need for the Cu2+ ion to maintain as high a coordination state as possible as water ligands are gradually lost. This is best achieved when it is located in the 6r due to the shorter O–Cu distances compared to the 8r.18 The deNOx conversion profiles (Fig. 1) match the 6r occupancy in both samples to a very high degree, particularly at low temperatures. Maximum NOx conversion is reached and is stable at 200 °C in Cu-SSZ-13-2, temperature at which the 6r occupancy considerably exceeds the 8r occupancy in the PXRD data. Cu-SSZ-13-1 in turn, exhibits a lower activity at this temperature, coincident with a lower fraction of Cu present in the 6r. The match is not exact since these results were obtained from two separate experiments and the presence of adsorbates (species involved in the catalysis) will influence the rate of Cu cation diffusion.7 However, as the data obtained under standard NH3-SCR conditions demonstrate, the majority of Cu species present are those located in the 6r. We can therefore conclude from this study that there is a strong correlation between Cu ions being present in the 6r and deNOx activity in the standard NH3-SCR reaction, especially at low temperatures.
The observation of a mixture of Cu sites in this study and by others at temperatures typically below 200 °C has rendered the determination of the active Cu species for NH3-SCR difficult. As has been shown previously, the Cu species in the vicinity of the 8r and 6r have different redox behaviour; the Cu ions present in the 8r have a lower coordination with the framework and are therefore easier to reduce than those in the 6r.8 One might imagine this to be important in the context of a catalytic process based on a redox mechanism. However, our results indicate that the presence of Cu ions in the 8r, even at moderate temperatures (∼200 °C) is lower than for the 6r and diminishes with increasing temperature, particularly in the highly active catalyst Cu-SSZ-13-2. This raises the question of whether Cu ions in the 8r are simply spectator species waiting to participate in the reaction? There is also some evidence, however, that low and high temperature regimes with different mechanisms may exist.1 In which case, our results suggest that Cu ions in the 8r, exhibiting high reducibility are active at low temperatures, whilst those in the 6r are active/more important at higher temperatures. In either case these results demonstrate that detailed structural analysis of high quality in situ measurements can be the key to understanding the active species in catalysts.
Notes and references
- A. M. Beale, F. Gao, I. Lezcano-Gonzalez, C. H. F. Peden and J. Szanyi, Chem. Soc. Rev., 2015, 44, 7371–7405 RSC.
- F. Gao, E. D. Walter, M. Kollar, Y. Wang, J. Szanyi and C. H. F. Peden, J. Catal., 2014, 319, 1–14 CrossRef CAS.
- S. Sato, Y. Yoshihiro, H. Yahiro, N. Mizuno and M. Iwamoto, Appl. Catal., 1991, 70, L1–L5 CrossRef CAS.
- E. Dooryhee, C. R. A. Catlow, J. W. Couves, P. J. Maddox, J. M. Thomas, G. N. Greaves, A. T. Steel and R. P. Townsend, J. Phys. Chem., 1991, 95, 4514–4521 CrossRef CAS.
- M. C. Dalconi, A. Alberti and G. Cruciani, J. Phys. Chem. B, 2003, 107, 12973–12980 CrossRef CAS.
- D. W. Fickel, J. M. Fedeyko and R. F. Lobo, J. Phys. Chem. C, 2010, 114, 1633–1640 CAS.
- J. H. Kwak, T. Varga, C. H. F. Peden, F. Gao, J. C. Hanson and J. Szanyi, J. Catal., 2014, 314, 83–93 CrossRef CAS.
- E. Borfecchia, K. A. Lomachenko, F. Giordanino, H. Falsig, P. Beato, A. V. Soldatov, S. Bordiga and C. Lamberti, Chem. Sci., 2015, 6, 548–563 RSC.
- J. Hun Kwak, H. Zhu, J. H. Lee, C. H. F. Peden and J. Szanyi, Chem. Commun., 2012, 48, 4758–4760 RSC.
- I. Lezcano-Gonzalez, U. Deka, B. Arstad, A. Van Yperen-De Deyne, K. Hemelsoet, M. Waroquier, V. Van Speybroeck, B. M. Weckhuysen and A. M. Beale, Phys. Chem. Chem. Phys., 2014, 16, 1639–1650 RSC.
- R. Martinez-Franco, M. Moliner, J. R. Thogersen and A. Corma, ChemCatChem, 2013, 5, 3316–3323 CrossRef CAS.
- C. W. Andersen, M. Bremholm, P. N. R. Vennestrom, A. B. Blichfeld, L. F. Lundegaard and B. B. Iversen, IUCrJ, 2014, 1, 382–386 CAS.
- I. Lezcano-Gonzalez, D. S. Wragg, W. A. Slawinski, K. Hemelsoet, A. Van Yperen-De Deyne, M. Waroquier, V. Van Speybroeck and A. M. Beale, J. Phys. Chem. C, 2015, 119, 24393–24403 CAS.
- U. Deka, A. Juhin, E. A. Eilertsen, H. Emerich, M. A. Green, S. T. Korhonen, B. M. Weckhuysen and A. M. Beale, J. Phys. Chem. C, 2012, 116, 4809–4818 CAS.
- G. W. Stinton and J. S. O. Evans, J. Appl. Crystallogr., 2007, 40, 87–95 CAS.
- M. Amri and R. I. Walton, Chem. Mater., 2009, 21, 3380–3390 CrossRef CAS.
- D. A. Woodcock, P. Lightfoot, P. A. Wright, L. A. Villaescusa, M. J. Diaz-Cabanas and M. A. Camblor, J. Mater. Chem., 1999, 9, 349–351 RSC.
- S. T. Korhonen, D. W. Fickel, R. F. Lobo, B. M. Weckhuysen and A. M. Beale, Chem. Commun., 2011, 47, 800–802 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc00513f |
| This journal is © The Royal Society of Chemistry 2016 |