
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Flexible macrocycles as versatile supports for catalytically active metal clusters†
Jason D.
Ryan
a,
Kevin J.
Gagnon
b,
Simon J.
Teat
b and
Ruaraidh D.
McIntosh
*a
aInstitute of Chemical Sciences, Heriot-Watt University, Riccarton, Edinburgh EH14 4AS, UK. E-mail: R.McIntosh@hw.ac.uk; Tel: +44 (0)131 451 8039
bLawrence Berkeley National Laboratory, 1 Cyclotron Road, MS 6R2100, Berkeley, California 94720, USA
First published on 12th February 2016
Abstract
Here we present three structurally diverse clusters stabilised by the same macrocyclic polyphenol; t-butylcalix[8]arene. This work demonstrates the range of conformations the flexible ligand is capable of adopting, highlighting its versatility in metal coordination. In addition, a Ti complex displays activity for the ring-opening polymerisation of lactide.
Transition metal nanoparticles often have properties quite distinct from those found in the bulk metal.1 This has a particular relevance for catalysis where decreasing the particle size obscures the boundary between reactivity at a surface and that of a molecular complex. Upon doing so we transition from heterogeneous to homogeneous catalysis. Very small particles (<50 Å) can be unstable (e.g. Ostwald ripening) and quickly aggregate to form larger species. Therefore, if we wish to create a continuum of sizes from monometallic complexes up to bulk metal we require a methodology to stabilise small particles and clusters. At the heart of cluster chemistry is a continued drive for controlled synthesis of targeted complexes. Supporting ligands are key to achieving this aim as they provide predictable metal binding sites, improve solubility and ultimately stabilise the resultant cluster. Calix[n]arenes are a family of macrocycles composed of n phenolic groups linked by methylene bridges. They are an attractive choice for supporting ligands as they are cheap to produce and can be modified extensively using well established methodology.2
The first Ti–calix[n]arene complexes were published independently by Power3 and Atwood4 (Fig. 1A and B respectively), more followed5 with recent reports detailing mixed metal complexes6 (e.g. Na/Ti species Fig. 1C6a). In all TBC[4] complexes common binding motifs persist with the Ti centre coordinated to all phenolate O atoms. Similarly, η4-coordination of TBC[4] to other high oxidation state transition/lanthanide metals is commonly observed.7 Several Ti–TBC[4] complexes display catalytic activity in diverse fields such as the ring opening polymerisation of lactide,8 aldol reactions9 and olefin epoxidation.10 It has been noted, in the case of multi-metallic species (i.e. those containing >1 Ti), that the complexes are stable in solution and increasing the number of metal centres has a positive effect on the catalytic reaction rate.9,11 Less studied are the larger calixarenes such as t-butylcalix[8]arene TBC[8]. To our knowledge the only reported Ti–TBC[8] complex is an anionic [Ti2(TBC[8])(OiPr)2]− species (Fig. 1D).12 Here, the flexibility of the ligand allows it to adopt a C-shaped, ‘tennis-ball’ conformation, allowing each Ti(IV) to coordinate to four of the eight phenolic oxygens. Synthesis involves initial treatment of the calixarene with base (alkali metal or hindered amine) followed by addition of the Ti reagent. The same anionic Ti complex is formed regardless of the base used.
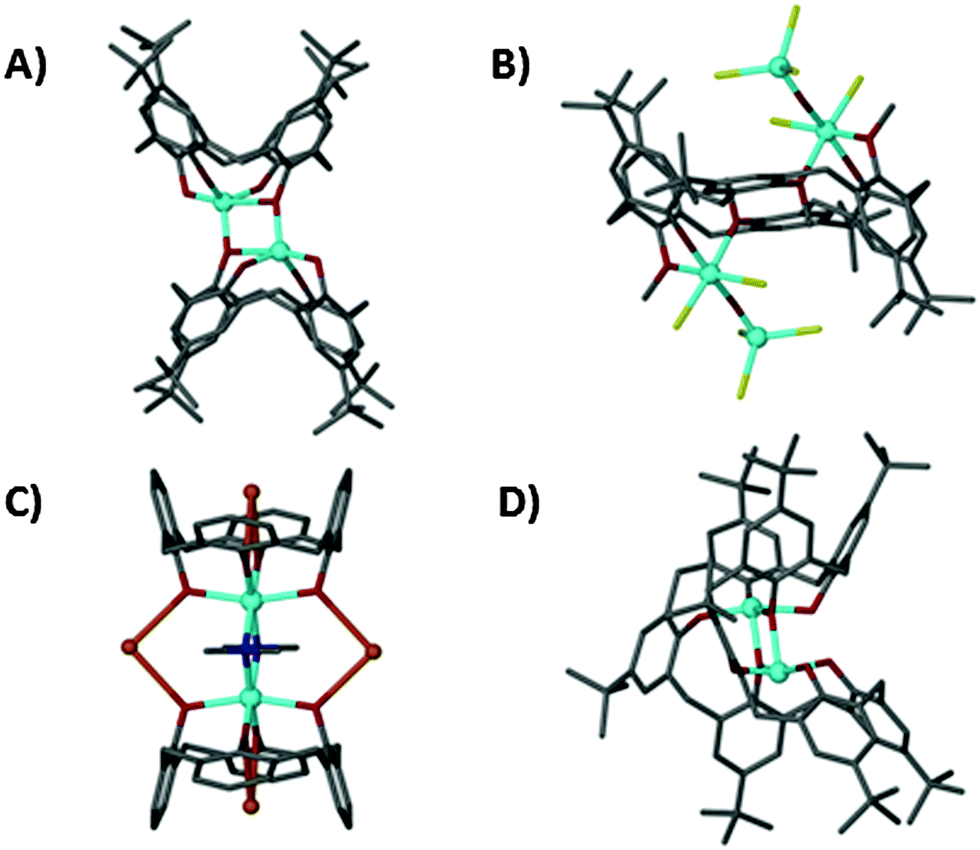 | ||
| Fig. 1 Examples of Ti(IV) calix[n]arene complexes from Power, Atwood, Raston and Pedersen (A–D respectively). Clockwise from top left: Ti2TBC[4]2 (A), Ti4TBC[6]Cl10 (B), Ti2Na2C[4]2 (C), [Ti2TBC[8](OiPr)2]− (D). Colour code: C = grey, O = red, Ti = blue, Cl = yellow, Na = orange. H-atoms omitted for clarity. | ||
With a view to creating metallic clusters we began to explore the coordination chemistry of TBC[8]. The large number of donor atoms and the larger macrocyclic annulus (cf. TBC[4]), increases the diversity of complex it could support. In addition, the variety of conformers the ligand can adopt13 (and the low barrier to their interconversion) offers scope to create unusual binding motifs. We speculate that this could restrict access to a catalytically active core, thus influencing reactivity and selectivity. Our initial studies focussed on a salt-elimination reaction whereby the phenolic protons would be removed with a strong base prior to the addition of TiX4. Treating TBC[8] with NaH in THF generated the anticipated anion due to deprotonation of the phenol/s. However, we noted no further reaction upon addition of TiCl4. To investigate why the trans-metallation step was unsuccessful we sought to gain insight through isolation of the intermediate Na complex. Eight equivalents of NaH were added to a THF solution of TBC[8] which, after two hours, was then concentrated and chilled. After several days at −15 °C, small yellow single crystals formed. These degraded quickly in air and an X-ray diffraction study was hampered by their weak scattering. Despite the relatively poor data quality, a neutral Na6TBC[8](THF)7 complex (Fig. 2, 1) was structurally elucidated. It is worth noting that the same complex is formed (albeit in lower yield) if fewer equivalents of NaH are added. Representative Na–O distances of 2.407(3) Å and 2.234(3) Å for Na1–O1 and Na3–O5 respectively are typical for Na phenolates.14 Stabilising Na–π interactions were observed between Na3–C34/C39 and Na5–C78/C83 (Fig. S1, ESI†). The Na to aryl centroid distances were calculated to be 2.586 Å and 2.540 Å, respectively, similar to those expected for alkali metal–π interactions. The calixarene is not fully deprotonated but remaining phenolic protons could not be located from the difference map.
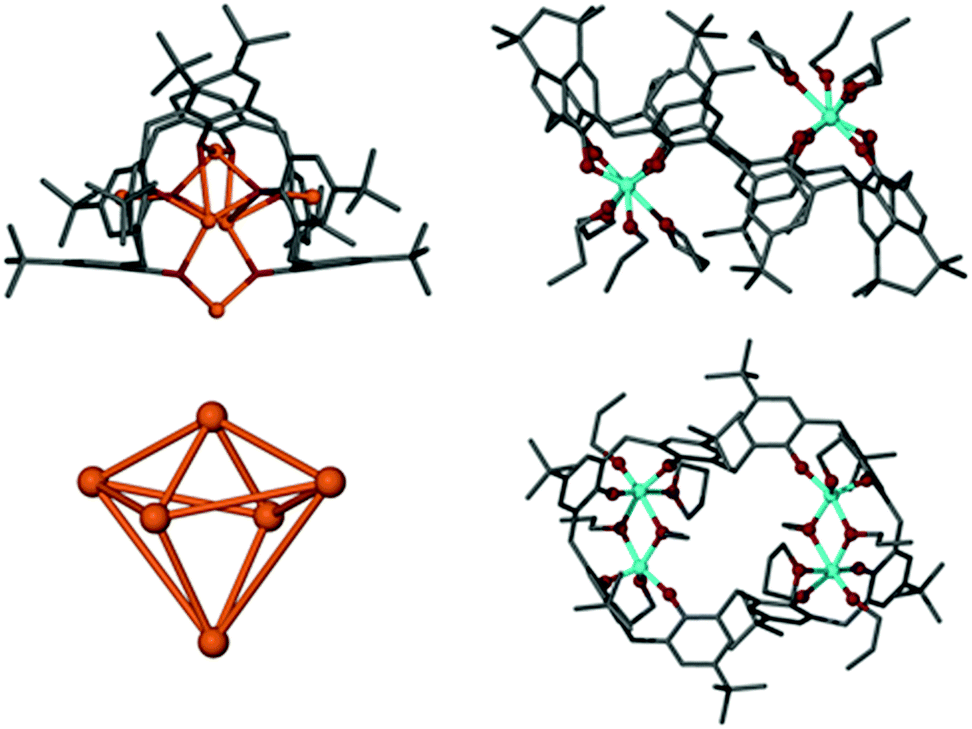 | ||
| Fig. 2 Top left: Na6TBC[8](THF)7 (1) complex and below, the Na6 metallic core. Top right: Ti4TBC[8](OnPr)8(THF)2 (2) and below, the orthogonal view. Colour code: C = grey, O = red, Ti = blue, Na = orange. H-atoms omitted for clarity. | ||
Unusually for alkali metal–calix[8]arene complexes,2 the ligand is tightly wrapped around the metallic core in a tennis-ball conformation (Fig. 3, 1), similar to that observed for Pedersen's Ti complex (Fig. 1D). It is plausible that access to the enclosed Na6 cluster is therefore restricted, explaining its reluctance to undergo trans-metallation. To bypass the unreactive Na species we elected to directly react the calixarene with Ti(OnPr)4. Four equivalents of Ti(OnPr)4 were added to a cloudy solution of TBC[8] in THF which turned orange/red after a few minutes. The mixture was concentrated under vacuum and slowly yielded red block-shaped crystals over several days at −15 °C. A single crystal X-ray diffraction study confirmed the formation of a neutral Ti complex; Ti4TBC[8](OnPr)8(THF)2 (Fig. 2, 2). Here TBC[8] adopts an unusual double-cone conformation (Fig. 3, 2) in which the two TBC[4]-like units are inverted with respect to one another.
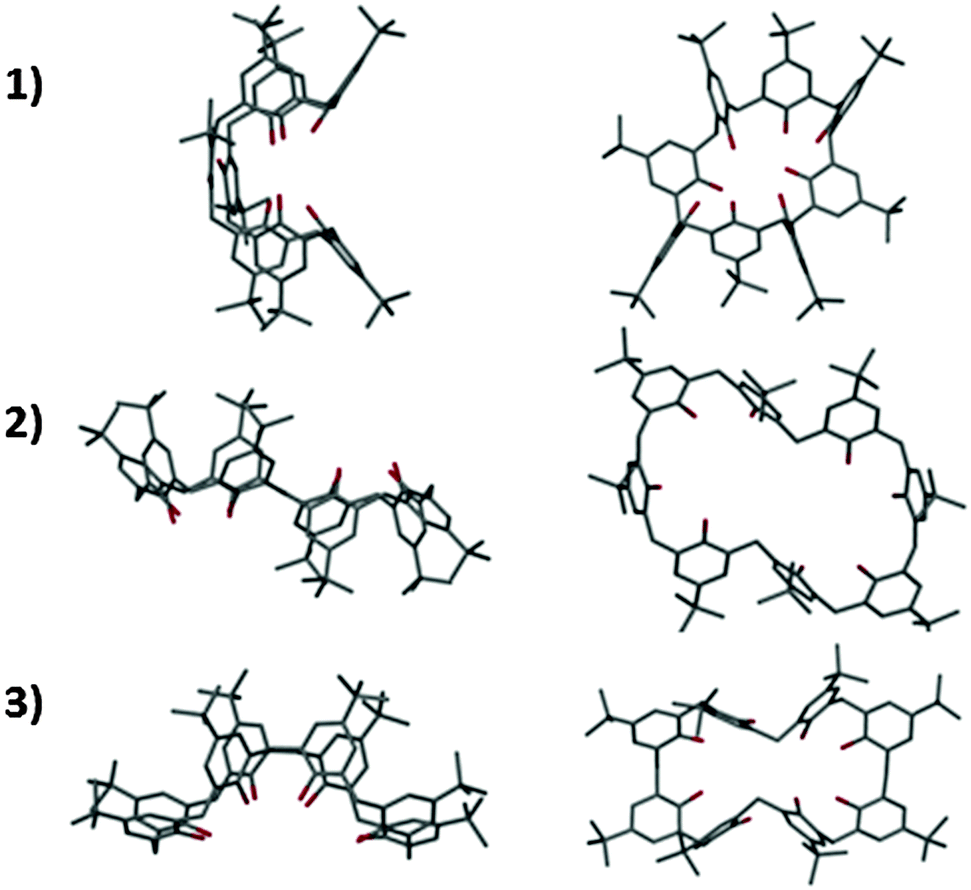 | ||
| Fig. 3 From top to bottom: orthogonal views (left and right) highlighting the conformation of the TBC[8] ligand in complexes 1–3. All H and non-calixarene atoms removed for clarity. Colour code: C = grey, O = red. | ||
Two Ti(IV) centres lie 3.2876(18) Å apart within each of the TBC[4] like units. The Ti2 units are supported by two bridging propoxides with Ti–O distances ranging from 1.969(5) Å to 2.113(4) Å. Each Ti is also coordinated to a terminal propoxide with shorter Ti–O bonds spanning from 1.801(5) Å to 1.807(5) Å. Ti(IV)-alkoxides are known to be active catalysts for the ring-opening polymerisation of lactide (LA).15 We predicted 2 may exhibit similar activity so undertook an investigative, proof-of-concept study. A flask containing 2 dissolved in toluene was charged with 100 equivalents of rac-LA before being placed in an oil bath heated to 130 °C. After sixteen hours the reaction was terminated and polymer precipitated by the addition of acidified MeOH. Mass spectral analysis indicates each polymer chain is terminated by –OPr (Fig. S2, ESI†). In general, Mw values are lower than anticipated with a maximum observed m/z of 1956. We attribute the absence of longer oligomers/polymers to intramolecular transesterification processes. This is evidenced by peaks in the mass spectrum separated by m/z equal to C3H4O2 (i.e. half a monomer unit) and signals in the 1H NMR spectrum at δ 5.29 (Fig. S3, ESI†). These are slightly higher than those expected for poly-LA (δ ∼ 5.1)16 and indicative of cyclic esters.17 A more thorough study of the polymerisation is currently underway but we propose the transesterification processes could be limited by reducing the reaction time.
Slow evaporation of 2, from deuterated acetone, yielded deep red single crystals after ten days at room temperature. The crystals were characterised by X-ray diffraction studies which exposed the instability of 2 through formation of a new species; Ti8TBC[8]2(μ2-O)2(μ3-O)6(OH)2(OnPr)2(acetone)2 (Fig. 4, 3). This complex consists of two fully deprotonated TBC[8] ligands in a double cone conformation (Fig. 3), this time with the TBC[4]-like units directed towards one another, the more commonly observed arrangement. A metallic core of eight Ti(IV) centres is bound to all sixteen of the TBC[8] phenoxides, eight μ-O atoms, two terminal hydroxides and two propoxides. The remaining coordination sites are occupied by ligated acetone molecules. We attribute the presence of μ-O to adventitious capture of water from the ‘wet’ deuterated solvent. Whilst there is little change to the gross stoichiometry of the complex there has clearly been a significant reorganisation of the Ti ions. Whilst we cannot currently postulate a mechanism for this process we do note that the Ti8 cluster is composed of two, identical and conjoined Ti4 units. These units, when viewed in isolation, bear a striking resemblance to the wing-tipped butterfly commonly observed for Mn–TBC[4] complexes (Fig. 5) reported by Dalgarno and Brechin.18 We tentatively suggest that formation of this favourable TM4 cluster motif is the driving force for the reorganisation and formation of 3.
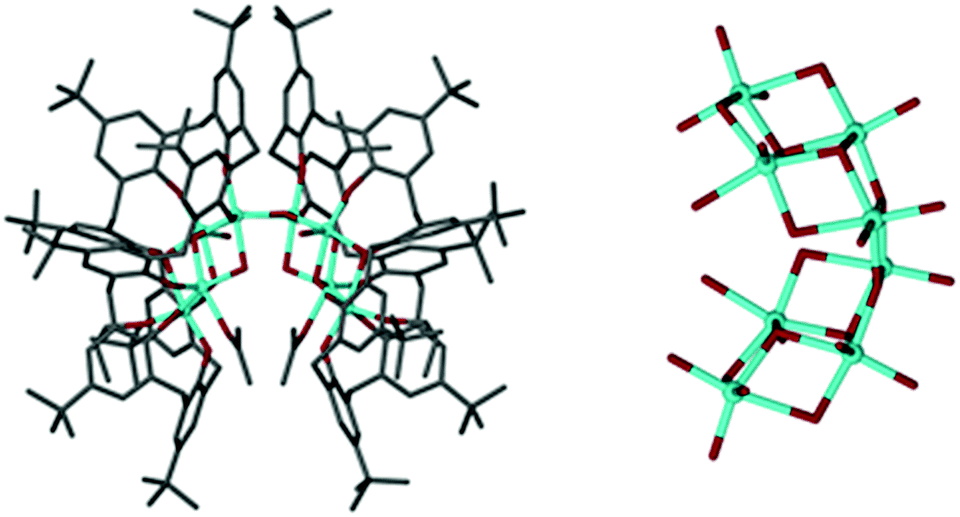 | ||
| Fig. 4 Ti8TBC[8]2(μ2-O)2(μ3-O)6(OH)2(OnPr)2(acetone)2 (3) and on the right an alternative view of its metallic core. Colour code: C = grey, O = red, Ti = blue. H-atoms omitted for clarity. | ||
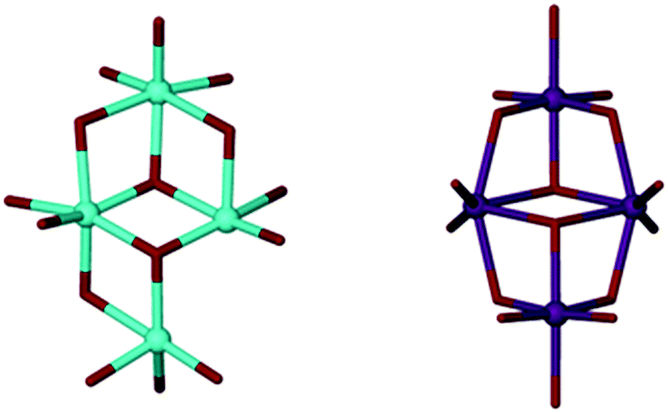 | ||
| Fig. 5 Left: One of the Ti4 sub-units which comprise the Ti8 cluster core of 3. Right: The wing-tipped butterfly core of an Mn4TBC[4](DMF)4 complex. Colour code O = red, Ti = blue, Mn = purple. H-atoms omitted for clarity. | ||
In summary, we have detailed the synthesis of several new TBC[8]-supported clusters. A methodological understanding has been gained over their synthesis as the obvious route of deprotonation followed by metallation/salt formation has been found to be ineffective due to formation of a stable (under Schlenk conditions) Na6 cluster. Preliminary investigation shows that although 2 does display activity as a ring-opening polymerisation catalyst for LA, the apparent instability of the complex highlights the need for a detailed study of the mechanism and elucidation of the active species. This will be the focus of future work and shall be reported in due course.
We are grateful to acknowledge financial support from Heriot-Watt University and assistance from the EPSRC UK National Mass Spectrometry Facility at Swansea University. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Notes and references
- For example: B. C. Gates, Chem. Rev., 1995, 95, 511 CrossRef CAS.
- C. D. Gutsche, Calixarenes 2001, Kluwer Academic Publishers, 2001 and references therein Search PubMed.
- M. M. Olmstead, G. Sigel, H. Hope, X. Xu and P. P. Power, J. Am. Chem. Soc., 1985, 107, 8087 CrossRef CAS.
- S. G. Bott, A. W. Coleman and J. Atwood, J. Chem. Soc., Chem. Commun., 1986, 610 RSC.
- For examples: A. Zanotti-Gerosa, E. Solari, L. Giannini, C. Floriani, N. Re, A. Chiesi-Villa and C. Rizzoli, Inorg. Chim. Acta, 1998, 270, 298 CrossRef CAS; W. Clegg, M. R. J. Elsegood, S. J. Teat, C. Redshaw and V. C. Gibson, J. Chem. Soc., Dalton Trans., 1998, 3037 RSC; S. R. Dubberly, A. Friedrich, D. A. Willman, P. Mountford and U. Radius, Chem. – Eur. J., 2003, 9, 3634 CrossRef PubMed.
- (a) A. J. Petrella, D. C. Craig, R. N. Lamb, C. L. Raston and N. K. Roberts, Dalton Trans., 2003, 4590 RSC; (b) A. J. Petrella, D. C. Craig, R. N. Lamb, C. L. Raston and N. K. Roberts, Dalton Trans., 2004, 327 RSC; (c) F. A. Cotton, E. V. Dikarev, C. A. Murillo and M. A. Petrukhina, Inorg. Chim. Acta, 2002, 32, 41 CrossRef.
- For examples: S. Sanz, K. Ferreira, R. D. McIntosh, S. J. Dalgarno and E. K. Brechin, Chem. Commun., 2011, 47, 9042 RSC; S. R. Dubberly, A. Friedrich, D. A. Willman, P. Mountford and U. Radius, Chem. – Eur. J., 2003, 9, 3634 CrossRef CAS PubMed; A. Caselli, E. Solari, R. Scopelliti, C. Floriani, N. Re, C. Rizzoli and A. Chiesi-Villa, J. Am. Chem. Soc., 2000, 122, 3652 CrossRef; J. A. Acho, L. H. Doerrer and S. Lippard, Inorg. Chem., 1995, 34, 2542 CrossRef.
- M. Frediani, D. Sémeril, D. Matt, L. Rosi, P. Frediani, F. Rizzolo and A. M. Papini, Int. J. Polym. Sci., 2010, 490724 Search PubMed.
- A. Soriente, M. De Rosa, M. Fruilo, L. Lepore, C. Gaeta and P. Neri, Adv. Synth. Catal., 2005, 347, 816 CrossRef CAS.
- J. M. Notestein, A. Solovyov, L. R. Andrini, F. G. Requejo, A. Katz and E. Iglesia, J. Am. Chem. Soc., 2007, 129, 15585 CrossRef CAS PubMed.
- D. M. Homden and C. Redshaw, Chem. Rev., 2008, 108, 5086 CrossRef CAS PubMed , and references therein.
- G. E. Hofmeister, F. E. Hann and S. F. Pedersen, J. Am. Chem. Soc., 1989, 111, 2318 CrossRef CAS.
- C. D. Gutsche and L. J. Bauer, J. Am. Chem. Soc., 1985, 107, 6052–6059 CrossRef CAS; C. D. Gutsche, Acc. Chem. Res., 1983, 16, 161 CrossRef.
- For a representative example see: C. Redshaw, D. Homden, D. L. Hughes, J. A. Wright and M. R. J. Elsegood, Dalton Trans., 2009, 1231 RSC.
- S. Gendler, S. Segal, I. Goldberg, Z. Goldschmidt and M. Kol, Inorg. Chem., 2006, 45, 4783 CrossRef CAS PubMed; Y. Kim, G. K. Jnaneshwara and J. G. Verkade, Inorg. Chem., 2003, 42, 1437 CrossRef PubMed; C. K. A. Gregson, V. C. Gibson, N. J. Long, E. L. Marshall, P. J. Oxford and A. J. P. White, J. Am. Chem. Soc., 2006, 128, 7410 CrossRef PubMed; C. Bakewell, G. Fateh-Iravani, D. W. Beh, D. Myers, S. Tabthong, P. Hormnirun, A. J. P. White, N. Long and C. K. Williams, Dalton Trans., 2015, 12326 RSC.
- M. H. Chisholm, S. S. Iyer, M. E. Matison, D. G. McCollum and M. Page, Chem. Commun., 1997, 1999 RSC.
- M. H. Chisholm, J. C. Gallucci and H. Yin, Dalton Trans., 2007, 4811 RSC.
- S. M. Taylor, G. Karotsis, R. D. McIntosh, S. Kennedy, S. J. Teat, C. M. Beavers, W. Wernsdorfer, S. Piligkos, S. J. Dalgarno and E. K. Brechin, Chem. – Eur. J., 2011, 17, 7521 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental detail, crystallographic information file (CIF) and additional diagrams. CCDC 1447632–1447634. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc00478d |
| This journal is © The Royal Society of Chemistry 2016 |