
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWell-defined silica-supported zirconium–imido complexes mediated heterogeneous imine metathesis†
Bilel
Hamzaoui
,
Jérémie D. A.
Pelletier
,
Edy
Abou-Hamad
and
Jean-Marie
Basset
*
King Abdullah University of Science and Technology (KAUST), KAUST Catalysis Center (KCC), Thuwal, 23955-6900, Saudi Arabia. E-mail: jeanmarie.basset@kaust.edu.sa
First published on 15th February 2016
Abstract
Upon prolonged thermal exposure under vacuum, a well-defined single-site surface species [(![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si–O–)Zr(NEt2)3] (1) evolves into an ethylimido complex [(Si–O–)Zr(
Si–O–)Zr(NEt2)3] (1) evolves into an ethylimido complex [(Si–O–)Zr(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NEt)NEt2] (2). Reactions of 2 with an imine substrate result in imido/imine (NRi, R: Et, Ph) exchange (metathesis) with the formation of [(Si–O–)Zr(NPh)NEt2] (3). Compounds 2 and 3 effectively catalyze imine/imine cross-metathesis and are thus considered as the first heterogeneous catalysts active for imine metathesis.
NEt)NEt2] (2). Reactions of 2 with an imine substrate result in imido/imine (NRi, R: Et, Ph) exchange (metathesis) with the formation of [(Si–O–)Zr(NPh)NEt2] (3). Compounds 2 and 3 effectively catalyze imine/imine cross-metathesis and are thus considered as the first heterogeneous catalysts active for imine metathesis.
Surface organometallic chemistry (SOMC) has allowed the access to a rich library of surface complexes active for a wide range of catalytic reactions. The nature of the fragments (M
C, M–C, M–H, M–NC) borne by the supported metal is pivotal to its reactivity toward targeted catalysis (e.g. alkane metathesis,1 alkene metathesis,2 methane nonoxidative coupling). Surface [M]NR fragments have not been well-investigated within this framework and their characterization and reactivity studies remain limited to date.
In the general context of both homogeneous and heterogeneous catalyses, transition metals forming multiple bonds with ligands are key in several industrially important chemical processes such as alkane oxidation3–5 (MO) or alkene metathesis6–8 (MCR2). In contrast, their metal imide (MNR) counterparts have received less attention yet can act as an intermediate in imido group (NR) transfer reactions.9 The importance of imido complexes and their isolable analogues is well-established in organic synthesis and catalysis.10,11 Stoichiometric or catalytic reactions may occur at the MNR fragment itself,12 or the imido group may remain a spectator, as in Schrock's group 6 olefin metathesis catalysts13 and imido-based Ziegler–Natta type olefin polymerisation catalysts.14 Catalytic imine metathesis is analogous to olefin metathesis in that two different imines afford a statistical mixture of all possible NR exchange products.
 | (1) |
NR)(THF) (Cp′ = Cp, Cp*) with imines.15–17 On the other hand, kinetic studies by Mountford and co-workers support an amine-catalyzed process to explain the reaction of (py)3Cl2Ti(NR) with imines.9,18
More recently, imine metathesis (eqn (1)) catalyzed by transition metals (Zr,15–17 Ti,9 Mo,19 Re,20 Ta,21 Nb4) was investigated in a number of research groups, most likely on account of the mechanistic analogy with the more famous and synthetically relevant olefin metathesis.22,23 To date, heterogeneous catalysts for imine metathesis have not been reported. Our recent foray into silica supported zirconium amido catalysts24,25 encouraged us to examine the formation and the reactivity of the related metal imido complexes. Note that MN fragments supported on silica have been documented only as spectator ligands for olefin metathesis molybdenum catalysts.26–30
Herein, we describe both the first catalytic imine metathesis reactions using silica supported imide–zirconium complexes. All surface species were characterized by solid state (SS) NMR experiments, elemental analysis and FT-IR spectroscopy.
The surface complex 1 was prepared by treating SiO2–700 with a solution of Zr(NEt2)4 in pentane (1.1 equivalents with respect to surface silanols). After workup, 1 was obtained as a yellow powder containing 2.7% Zr, 4.3% C and 1.3% N with a Zr/C/N molar ratio of 1/12/3.1 (±0.3) (expected 1/12/3) and thus suggests a monopodal surface complex with three NEt2 ligands. Comparison of the FT-IR spectra of SiO2–700 and 1 (Fig. 1) is consistent with the chemical grafting of Zr(NEt2)4 onto the silica surface.
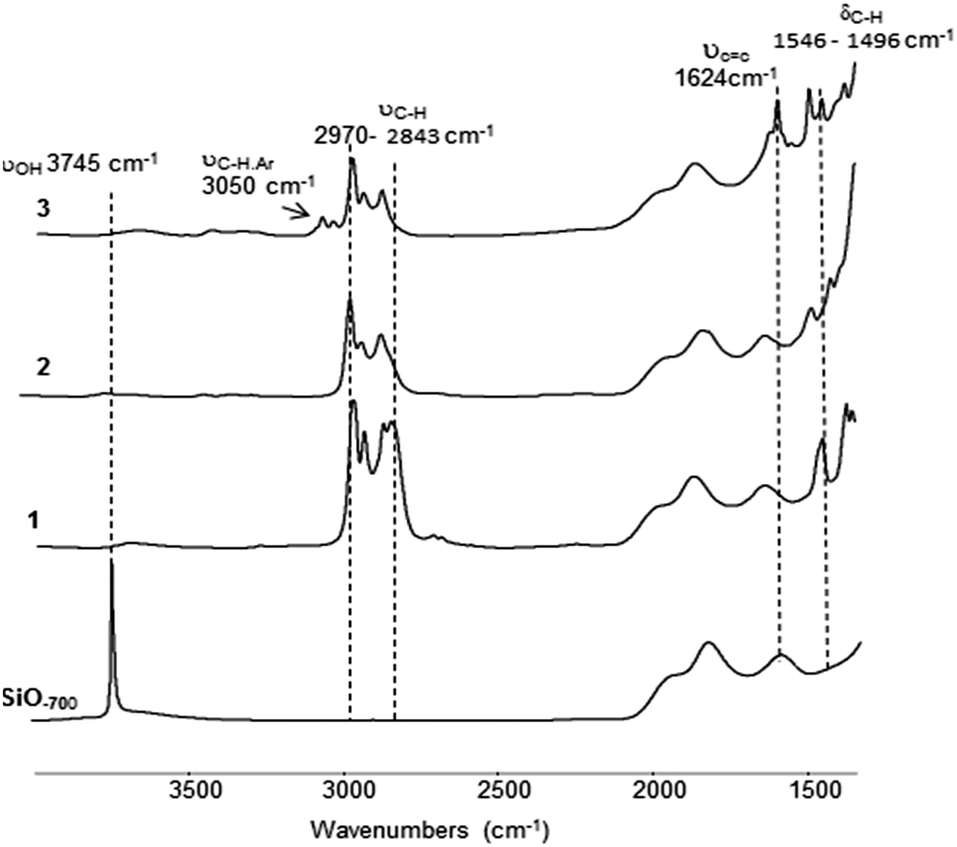 | ||
| Fig. 1 FT-IR of SiO2–700, 1 [Si–O–Zr(NEt2)3], 2 [Si–O–Zr(NEt)NEt2] and 3 [Si–O–Zr(NPh)NEt2]. | ||
The IR peaks associated with the isolated surface silanols (3745 cm−1) completely disappeared. Conversely, new IR bands associated with ν(CH) and δ(CH) of the diethylamine ligands are observed in the 2970–2843 cm−1 and 1546–1496 cm−1 regions, respectively.
All these observations and chemical analyses are consistent with the proposed structure of 1 (Fig. 2), but further corroborations at the molecular level have been sought using SS NMR spectroscopy of the grafted species. The solid-state 1H NMR spectrum of 1 shows two broad resonances at 1.1 and 3.2 ppm (Fig. 2A) and its 13C solid-state cross polarization/magic angle spinning (CP/MAS) NMR spectrum displays strong signals at 14 and 43 ppm (Fig. 2B).25
 | ||
| Fig. 2 (A) 1D 1H spin-echo MAS solid state NMR spectrum of 2, (B) 13C CP/MAS NMR spectrum of 2. (C) 2D 1H–1H double-quantum (DQ)/single-quantum (SQ), (D) 1H–1H triple-quantum (TQ)/SQ and (E). | ||
Moreover, the 2D HETCOR SS NMR spectra revealed the correlations between only pairs of attached 1H–13C spins using a short contact time of 0.5 ms. Clear correlations were noticeable between the protons at 1.1 and 3.2 ppm and the carbons at 14 and 43 ppm, respectively (Fig. 2E). Two-dimensional proton double- (DQ) and triple-quantum (TQ) correlation experiments were applied to determine the number of protons attached to the same carbon, hence discriminating between CH2 and CH3 groups. A strong autocorrelation peak is observed for the proton resonances at 1.1 ppm in both DQ and TQ spectra (Fig. 2C and D). This is compatible with the assignment of this resonance to methyl protons. The resonances at 3.2 ppm show a correlation at 6.4 ppm (ω1) in the DQ spectrum, but no autocorrelation peak is observed in the TQ spectrum. It can be thus assigned to a CH2 group. Furthermore, a correlation in the DQ NMR spectrum between the CH3 group at 1.1 ppm and the CH2 group at 3.2 ppm confirms that both groups are in proximity to each other. In conclusion, these data validate 1 as a surface zirconium complex exhibiting three equivalent diethyl-amido ligands [(Si–O–)Zr(NEt2)3].
The formation of imido ligands during the synthesis of molecular dialkylamido metal complexes is known to occur after thermal treatment under vacuum.31 The release of gaseous alkenes and dialkylamines has also been detected upon thermal transformation of isolable dialkylamido32 and imine complexes of the early transition metals.33 We decided to monitor the thermal treatment of 1 to observe possibly the conversion to zirconium imido surface species (Scheme 1).
 | ||
| Scheme 1 Silica-supported zirconium imido formation with the hypothetical intermediate.34 | ||
The initial evacuation of 1 at room temperature left the IR spectrum unchanged, although evacuation at elevated temperature (200 °C) resulted in the modification of the signal pattern (Fig. 1) concurrently to an overall reduction of the signal's intensity in the alkyl vibration region. Elemental analysis of 2 gives 2.7% Zr, 2.3% C and 0.9% N with a Zr/C/N ratio of 1/6.4/2.1 (±0.3) (expected 1/6/2). The volatiles identified and quantified by GC consist exclusively of diethylamine and ethylene (1/0.9). The 1H SS NMR spectrum of 2 contains two broad signals centered at 1.1 and 3.1 ppm (Fig. S1, ESI†). The 13C SS NMR spectrum of 2 (Fig. S2, ESI†) exhibits two major signals at 14.2 and 40.6 ppm and two minor signals at 18.7 and 36.2 ppm. The most intense peaks at 14.2 and 40.2 ppm are reminiscent of that previously seen for 1 at 14 ppm and 42.7 ppm. They were assigned to the carbon atoms of amino-ethyl groups in the N (CH2CH3)2 ligands. The other two peaks at 18.7 and 36.2 ppm were consistent with an N-ethyl group different to that already present on the zirconium center. Considering all the data, we can attribute them to the methyl and the methylene group borne by the nitrogen of an ethylimido ligand (NCH2CH3). All these results allow us to identify 2 as [(Si–O–)Zr(NEt)NEt2]. To the knowledge of the authors, this is the first report of a well-defined single site zirconium imido species supported onto silica.
Previous reports mainly described two types of NR transfer reactions: stoichiometric and catalytic. The former passes by imide/imine stoichiometric metathesis of a metal imide with one molecule of the imine substrate and is regiospecific. New metal imide and imine products are formed by a transient 4-membered metallacycle (eqn (2)).
 | (2) |
C)) (Fig. 1). A comparison of the FT-IR spectra of 2 and 3 reveals a notable decrease of ν(CH) (2970–2843 cm−1). Elemental analysis of 3 gives 2.7% Zr, 3.7% C and 0.95% N with a Zr/C/N ratio of 1/10.3/2.2 (±0.3) (expected 1/10/2). This is in agreement with one NEt2 ligand in 2 being replaced by an arylimido ligand.
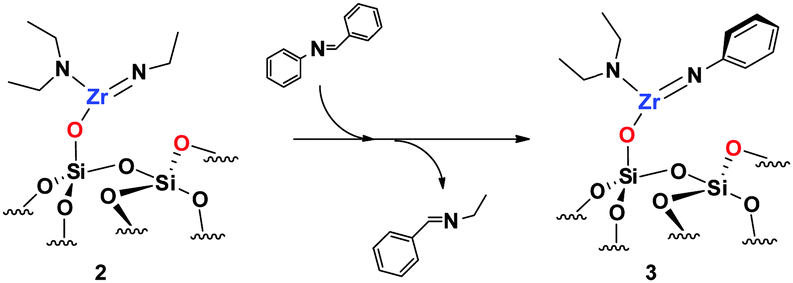 | ||
| Scheme 2 Silica-supported imide/imine metathesis. | ||
The 1H NMR spectrum of 3 contains an intense signal at 7.1 ppm (Fig. 3A), absent for 1 and 2, in addition to two overlapping peaks (a major one at 1.1 ppm and a minor one at 3 ppm). The 13C MAS spectrum displays also two new signals at 118 and 128 ppm consistent with aromatic carbon in addition to two signals at 14 ppm and 40 ppm (Fig. 3C). A correlation was observed in the HETCOR spectrum between the 1H signal at 7.1 ppm and 13C signals at 118 and 128 ppm (Fig. 3D). Moreover, the DQ experiment shows a double autocorrelation for the 7.1 ppm resonance as expected for an aromatic CH proton (Fig. 3B). These observations are in line with the presence of an aromatic group in 3. In the double-quantum (DQ) spectrum (Fig. 3B), the 1H signals at 1.1 and 3.0 ppm display strong auto-correlations that can be assigned to the NEt2 group similarly to that observed in 1.
 | ||
| Fig. 3 (A) 1D 1H spin-echo MAS solid state NMR spectrum of 3, (B) 2D 1H–1H double-quantum (DQ), (C) 13C CP/MAS NMR spectrum of 3, (D) 2D CP/MAS HETCOR NMR spectrum of 3 (see the ESI† for details). | ||
Species 3 was investigated as a catalyst for the imine metathesis reaction. Selected imine substrates were tested. All runs were carried out under similar conditions (80 °C, 4 mol% catalyst, toluene (0.4 ml)) and terminated after 2, 4 and 12 h (Table S1, ESI†). The products monitored by gas chromatography-mass spectrometry were consistent with those obtained in cross metathesis reaction (eqn (3)).
Reactions using imines such as N-(3,4-methylenedioxybenzylidene)benzylamine (entry 1) and benzal(benzhydryl)amine (entry 2) occurred in high yields (around 70%). The use of phenyl-(1-phenylethylidene) (entry 3) and N-trimethylsilylbenzaldimine (entries 4) resulted in lower yields (around 50% after 12 h) (Table 1). When species 2 was employed as a catalyst for the imine metathesis reaction in entry 5, the obtained yield was comparable to the same reaction using 3 as a catalyst in entry 2 (around 65% after 12 h).
 | (3) |




The conversions for both substrates were similar and the corresponding products were obtained with around the same yield values. Product pairs 6 and 7 are obtained with the same selectivities (50–50%) and no other products were observed.
Additional experiments were performed to test the zirconium system reusability. After 12 h, the catalytic material was isolated by filtration, washed with toluene, dried under high vacuum, and reused in imine metathesis (entry 1). This sequence was performed five times with only a slight loss of catalytic activity being observed (yield of 76% in the first cycle vs. 72% in the fifth cycle) (Fig. S3, ESI†). FT-IR of the catalysts after reaction was conducted which indicates that the catalytically active species are still present and no adsorbed products remain on the surface (Fig. S4, ESI†).
In our investigation of the reactions of Zr(NEt2)4 with the silica surface to give [(Si–O–)Zr(NEt2)3] (1), thermal treatment of 1 generated the imido complex [(Si–O–)Zr(NR)NEt2] (2). 2 can be converted to [(Si–O–)Zr(NPh)NEt2] (3) by imine metathesis using ArCNPh. Both 2 and 3 were tested as catalysts for imine metathesis using several imines and exhibited conversion ranging from 49 to 76%. 2 and 3 are the first reported heterogeneous catalysts for imine metathesis.
References
- E. Callens, N. Riache, K. Talbi and J. M. Basset, Chem. Commun., 2015, 51, 15300–15303 RSC.
- N. Riache, E. Callens, J. Espinas, A. Dery, M. K. Samantaray, R. Dey and J. M. Basset, Catal.: Sci. Technol., 2015, 5, 280–285 CAS.
- L. K. Woo, Chem. Rev., 1993, 93, 1125–1136 CrossRef CAS.
- J. W. Bruno and X. J. Li, Organometallics, 2000, 19, 4672–4674 CrossRef CAS.
- M. Sun, J. Z. Zhang, P. Putaj, V. Caps, F. Lefebvre, J. Pelletier and J. M. Basset, Chem. Rev., 2014, 114, 981–1019 CrossRef CAS PubMed.
- A. Bokka, Y. D. Hua, A. S. Berlin and J. Jeon, ACS Catal., 2015, 5, 3189–3195 CrossRef CAS.
- C. A. Denard, M. J. Bartlett, Y. J. Wang, L. Lu, J. F. Hartwig and H. M. Zhao, ACS Catal., 2015, 5, 3817–3822 CrossRef CAS.
- J. I. du Toit, C. G. C. E. van Sittert and H. C. M. Vosloo, Monatsh. Chem., 2015, 146, 1115–1129 CrossRef CAS.
- J. M. McInnes and P. Mountford, Chem. Commun., 1998, 1669–1670 RSC.
- V. C. Gibson, Adv. Mater., 1994, 6, 37–42 CrossRef CAS.
- W. A. Nugent and B. L. Haymore, Coord. Chem. Rev., 1980, 31, 123–175 CrossRef CAS.
- A. P. Duncan and R. G. Bergman, Chem. Rec., 2002, 2, 431–445 CrossRef CAS PubMed.
- R. R. Schrock and A. H. Hoveyda, Angew. Chem., Int. Ed., 2003, 42, 4592–4633 CrossRef CAS PubMed.
- P. D. Bolton and P. Mountford, Adv. Synth. Catal., 2005, 347, 355–366 CrossRef CAS.
- R. L. Zuckerman, S. W. Krska and R. G. Bergman, J. Am. Chem. Soc., 2000, 122, 751–761 CrossRef CAS PubMed.
- K. E. Meyer, P. J. Walsh and R. G. Bergman, J. Am. Chem. Soc., 1994, 116, 2669–2670 CrossRef CAS.
- K. E. Meyer, P. J. Walsh and R. G. Bergman, J. Am. Chem. Soc., 1995, 117, 974–985 CrossRef CAS.
- P. Mountford, Chem. Commun., 1997, 2127–2134 RSC.
- G. K. Cantrell and T. Y. Meyer, Organometallics, 1997, 16, 5381–5383 CrossRef CAS.
- W. D. Wang and J. H. Espenson, Organometallics, 1999, 18, 5170–5175 CrossRef CAS.
- M. C. Burland, T. W. Pontz and T. Y. Meyer, Organometallics, 2002, 21, 1933–1941 CrossRef CAS.
- J. W. Nelson, L. M. Grundy, Y. F. Dang, Z. X. Wang and X. T. Wang, Organometallics, 2014, 33, 4290–4294 CrossRef CAS.
- J. M. Bates, J. A. M. Lummiss, G. A. Bailey and D. E. Fogg, ACS Catal., 2014, 4, 2387–2394 CrossRef CAS.
- B. Hamzaoui, M. El Eter, E. Abou-Hamad, Y. Chen, J. D. A. Pelletier and J. M. Basset, Chem. – Eur. J., 2015, 21, 4294–4299 CrossRef CAS PubMed.
- B. Hamzaoui, J. D. A. Pelletier, M. El Eter, Y. Chen, E. Abou-Hamad and J. M. Basset, Adv. Synth. Catal., 2015, 357, 3148–3154 CrossRef CAS.
- P. Avenier, A. Lesage, M. Taoufik, A. Baudouin, A. De Mallmann, S. Fiddy, M. Vautier, L. Veyre, J. M. Basset, L. Emsley and E. A. Quadrelli, J. Am. Chem. Soc., 2007, 129, 176–186 CrossRef CAS PubMed.
- P. Avenier, X. Solans-Monfort, L. Veyre, F. Renili, J. M. Basset, O. Eisenstein, M. Taoufik and E. A. Quadrelli, Top. Catal., 2009, 52, 1482–1491 CrossRef CAS.
- F. Blanc, C. Coperet, J. Thivolle-Cazat and J. M. Basset, Angew. Chem., Int. Ed., 2006, 45, 6201–6203 CrossRef CAS PubMed.
- F. Blanc, N. Rendon, R. Berthoud, J. M. Basset, C. Coperet, Z. J. Tonzetich and R. R. Schrock, Dalton Trans., 2008, 3156–3158 RSC.
- F. Blanc, J. Thivolle-Cazat, J. M. Basset, C. Coperet, A. S. Hock, Z. J. Tonzetich and R. R. Schrock, J. Am. Chem. Soc., 2007, 129, 1044–1045 CrossRef CAS PubMed.
- D. C. Bradley and I. M. Thomas, Can. J. Chem., 1962, 40, 1355–1360 CrossRef CAS.
- K. J. Ahmed, M. H. Chisholm, K. Folting and J. C. Huffman, J. Am. Chem. Soc., 1986, 108, 989–999 CrossRef CAS.
- M. Beaudoin and S. L. Scott, Organometallics, 2001, 20, 237–239 CrossRef CAS.
- M. El Eter, B. Hamzaoui, E. Abou-Hamad, J. D. A. Pelletier and J. M. Basset, Chem. Commun., 2013, 49, 4616–4618 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, additional data, and figures and tables. See DOI: 10.1039/c6cc00471g |
| This journal is © The Royal Society of Chemistry 2016 |