
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
PIM-1 mixed matrix membranes for gas separations using cost-effective hypercrosslinked nanoparticle fillers†
Tamoghna
Mitra
a,
Rupesh S.
Bhavsar
b,
Dave J.
Adams
a,
Peter M.
Budd
*b and
Andrew I.
Cooper
*a
aDepartment of Chemistry, University of Liverpool, Brownlow Hill, Liverpool, Merseyside L69 7ZD, UK. E-mail: aicooper@liverpool.ac.uk
bSchool of Chemistry, University of Manchester, Manchester M13 9PL, UK. E-mail: Peter.Budd@manchester.ac.uk
First published on 21st March 2016
Abstract
High-free-volume glassy polymers, such as polymers of intrinsic microporosity (PIMs) and poly(trimethylsilylpropyne), have attracted attention as membrane materials due to their high permeability. However, loss of free volume over time, or aging, limits their applicability. Introduction of a secondary filler phase can reduce this aging but either cost or instability rules out scale up for many fillers. Here, we report a cheap, acid-tolerant, nanoparticulate hypercrosslinked polymer ‘sponge’ as an alternative filler. On adding the filler, permeability is enhanced and aging is strongly retarded. This is accompanied by a CO2/N2 selectivity that increases over time, surpassing the Robeson upper bound.
Cheap, energy-efficient separation processes are essential for industrial processes such as natural gas purification, hydrocarbon purification, hydrogen recovery, and post-combustion carbon capture.1–5 In recent years, polymer membranes have emerged as attractive candidates for separations because they are readily processed and potentially scalable.6,7 Polymer membranes usually separate gases by preferential permeation under a pressure gradient. However, fast permeation can generally only be achieved by compromising selectivity. Robeson et al.8 described this trade-off relationship using a double-logarithmic plot of selectivity vs. permeability, with the so-called ‘upper bound’ being the line on this plot described by the most effective membranes.
High-free-volume glassy polymers, such as polymers of intrinsic microporosity (PIMs) or poly[1-(trimethylsilyl)-1-propyne] (PTMSP), are highly permeable to gases like CO2 and demonstrate reasonable gas selectivity, making them attractive materials for gas separation membranes. The awkwardly shaped, bulky backbones of these polymers do not allow efficient packing of polymer chains, leaving fractional free volume to be exploited as gas transport pathways.9–12 However, over time, these polymer chains relax towards a more thermodynamically stable state at the expense of fractional free volume. As the fractional free volume contracts, gas transport pathways are reduced, leading to a loss of permeability.13
This physical aging process poses a major challenge for the commercial application of glassy polymers, and this has motivated a large body of research with the goal of ‘freezing in’ the free volume. Approaches include rigidifying the initial polymer structure using surface plasma treatment,14 co-polymerization,15 or crosslinking.16,17 These methods have been successful at reducing aging, but often with a major loss in permeability. Alternatively, a secondary phase, very often another porous material such as a metal–organic framework (MOF), a zeolite, or a porous organic cage, can be incorporated to form a mixed matrix membrane (MMM);18–20 these composite membranes can outperform the unfilled polymer and even surpass the Robeson upper bound performance.
Recently, it was reported that addition of a polymeric filler can reduce aging while improving selectivity many fold.21–23 This filler was a microporous organic polymer, PAF-1, which was incorporated at a loading of 10 wt%. While highly effective, there are some drawbacks to this polymer: it is synthesised from relatively expensive building blocks and requires air- and moisture-sensitive organometallic coupling agents, casting doubts on its scalability. Here we explore a water- and acid-tolerant hypercrosslinked polymer, or HCP,24 which is synthesised from cheap starting materials using relatively inexpensive and air-tolerant catalysts.
MMMs were formed by adding hypercrosslinked polystyrene (hereafter referred as HCP) as a filler into PIM-1 (Fig. 1). Most porous organic HCPs are prepared as monoliths or as aggregated powders.24,25 These polymers are difficult to grind post-synthesis to give submicron sized particles. We also found that adding irregular and large HCP filler particles tends to leave large polymer/particle interfacial defects, providing non-selective diffusion pathways and lowering selectivity (Fig. S7d, ESI†). We therefore prepared the filler particles using an emulsion polymerisation process. Our strategy was to prepare submicron-sized filler particles to ensure a homogeneous distribution of particles throughout the PIM-1. In this context, it should be mentioned that other very recent work has also demonstrated the use of HCP as filler, in that case with PTMSP as the continuous phase.26
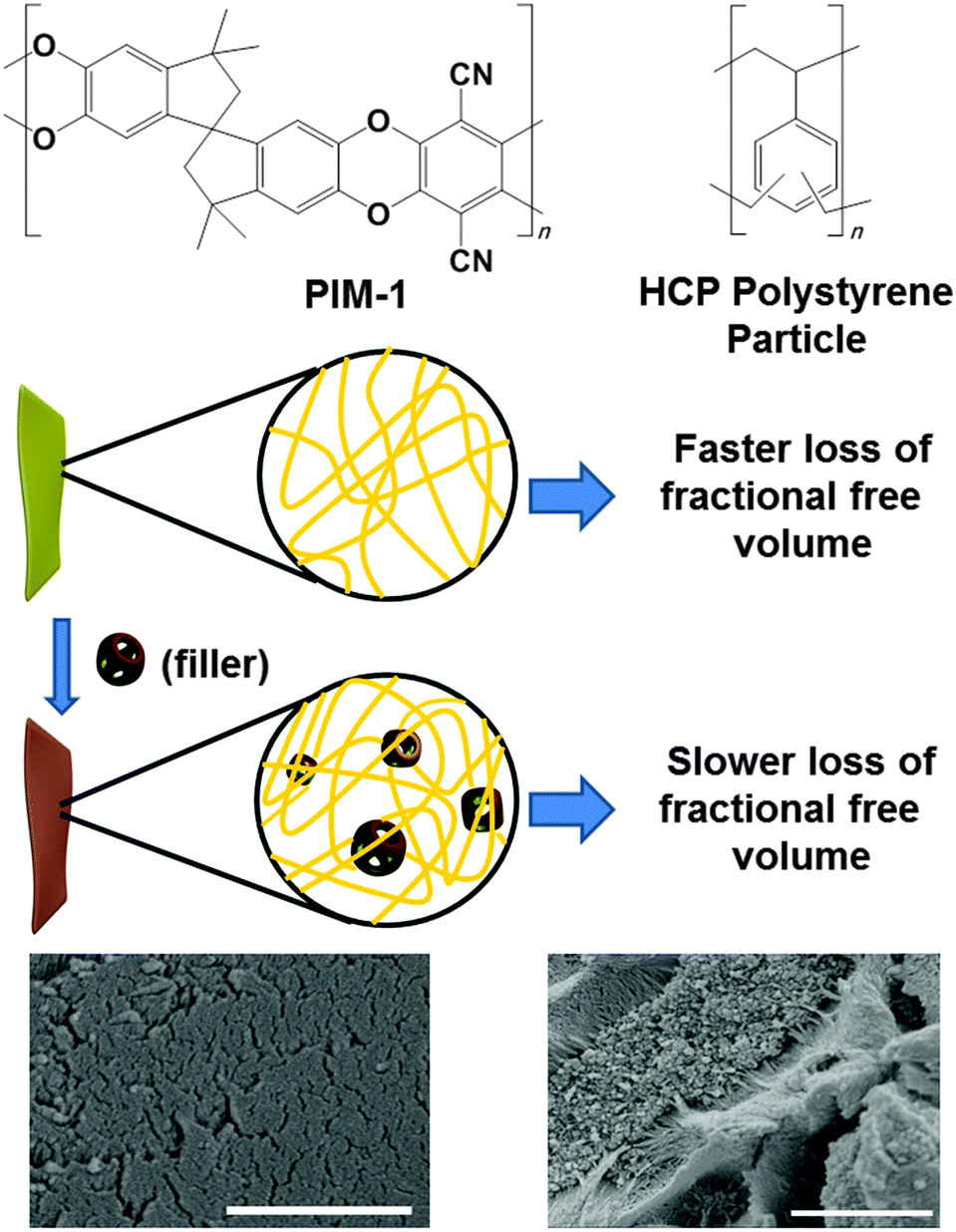 | ||
| Fig. 1 (top) Molecular structure of PIM-1 and the hypercrosslinked polymer used as the filler. (middle) Schematic illustration of effect of filler on overall performance of MMMs. SEM images of (bottom left) pristine PIM-1 and (bottom right) MMM with 16.67 wt% filler. Scale 1 μm. | ||
The filler HCP was synthesised by a previously reported method27 where a Friedel–Crafts hypercrosslinking reaction was used to crosslink preformed poly(vinyl benzyl chloride) particles (Fig. 1, top). The poly(vinyl benzyl chloride) particles were synthesised using emulsion polymerisation and hence the particle size could be controlled. The HCP had an average spherical particle diameter of approximately 55 nm (Fig. S2, ESI†), with a Brunauer–Emmett–Teller (BET) surface area of approximately 1700 m2 g−1 (Fig. S3, ESI†).27
MMMs of PIM-1 incorporating HCP were prepared by dispersing the HCP particles in a solution of PIM-1, in either chloroform or dichloromethane (DCM) (Fig. 1, bottom). After stirring for 20 hours to ensure effective mixing, the films were prepared by casting on a glass or Teflon mould. Due to the small and homogeneous particle size distribution of the filler, good dispersion of the two phases, PIM-1 and HCP, were achieved in these nanocomposite MMMs. A number of polymer-to-filler weight ratios were investigated.
Scanning electron microscopy (SEM) of the PIM-1/HCP MMMs showed spherical particles of filler embedded within the membrane (Fig. 1 and Fig. S7a, b, ESI†). A fibrous nanostructure of PIM-1 was also observed in the SEM images of all PIM-1/HCP membranes, which was not visible in pristine PIM-1 membranes. This may indicate phase separation in the presence of the filler particle. However, we suggest that this nanostructure should not compromise the selectivity/permeability of the membrane, due to more dense packing of nano-sized filler and PIM-1, compared to micron-sized filler. Visual inspections of the MMMs showed a reasonably homogeneous distribution of the HCP filler (Fig. S5 and S6, ESI†). The homogeneity of these membranes was also tested by area mapping with IR spectroscopy. A qualitative demonstration of the uniformity of both PIM-1 and filler across the area of the membrane was achieved by measuring IR spectra and comparing the area under the peak at the –CN stretching frequency (Fig. S4–S6, ESI†).
Single gas permeation data were obtained for all MMMs ‘as cast’ and after ethanol treatment (Table S1 and S2, ESI†). Permeability coefficients, P, were determined for N2 and CO2, and subsequently selectivities for the gas pair CO2/N2 at ambient temperature (∼298 K) were calculated. The ideal selectivity for a pair of gases, A and B, is the ratio of permeabilities, α(A/B) = PA/PB. Comparing the MMMs with different loadings of HCP filler (Fig. 2), it is clear that permeability increases with filler content. This can be understood by comparing the BET surface area of the filler (∼1700 m2 g−1) and the BET surface area of PIM-1 (∼750–850 m2 g−1). Since the surface area of the PIM-1 is about half that of the filler and the permeability is averaged over both the filler and matrix phase, the permeability increases with increasing filler loading. Permeability with regards to CO2 could be increased up to ∼250% compared to PIM-1 alone.
 | ||
| Fig. 2 Dependence of permeability coefficient of CO2, (top), and ideal selectivity relative to N2, (bottom). (-◇-): cast from DCM followed by ethanol treatment; (-△-): cast from chloroform followed by ethanol treatment. All data are shown for non-aged membranes. | ||
Whilst the permeability of PIM-1/HCP-Polystyrene membrane increases dramatically on addition of the filler, there is also a significant drop in the selectivity on addition of 5 wt% filler. However, from 5 to 15 wt% filler, there was little further drop in selectivity, despite the increase in permeability over this filler range. At higher amounts of filler (>15 wt%), the selectivity is compromised. This is in accordance with Robeson's8 observation that permeability is generally increased at a cost of selectivity.
For the comparison shown in Fig. 2, the membranes were treated with ethanol. It is well known that the permeability performances of PIM-1 membranes may vary based on the casting conditions (for example, the choice of solvents). However, alcohol can wash away residual casting solvent and such alcohol treatment provides a means for comparisons between different membranes.28 Alcohol treatment also helps swell the membrane, opening up free volume. Hence, an alcohol wash increases the permeability dramatically,28 although at the expense of selectivity. In this context, it is important to note that the HCP particles do not swell much in presence of ethanol (Fig. S11, ESI†). Hence, this additional gain of permeability can be attributed to swelling of PIM-1.
Upon aging, relaxation of the PIM-1 chains in ethanol treated membranes leads to a faster loss of permeability than for untreated membranes (Fig. 3). The as-cast MMMs (using DCM as the casting solvent) show relatively little loss of CO2 permeability (9.4–20.1% after 150 days) while the same membranes after ethanol treatment show a loss of permeability towards CO2 of up to 28% over the same time period. However, the membranes cast from chloroform show faster aging, losing up to 50% permeability towards CO2. Once ethanol-treated, both MMMs show very similar behaviour and trends. Fig. 3 and Fig. S9 (ESI†) summarise the aging properties of each membrane. In most cases, loss of N2 permeability due to aging is significantly faster than the loss of CO2 permeability. Hence, the performance of all MMMs with regards to CO2/N2 selectivity increases over time. In some cases, the membranes show a performance that exceeds the 2008 Robeson upper bound (Fig. 4). Additionally, incorporation of the filler significantly retards the decrease in CO2 permeability as compared to PIM-1 alone.
 | ||
Fig. 3 Permeability and aging characteristic of MMMs with HCP polystyrene as filler. Top: Cast from DCM and subsequent treatment by ethanol bottom: cast from chloroform and subsequent treatment by ethanol. Colour code: (■) N2 permeability on first day; ( ) N2 permeability on 150th day; ( ) N2 permeability on 150th day; ( ) CO2 permeability on first day, ( ) CO2 permeability on first day, ( ) CO2 permeability on 150th day. ) CO2 permeability on 150th day. | ||
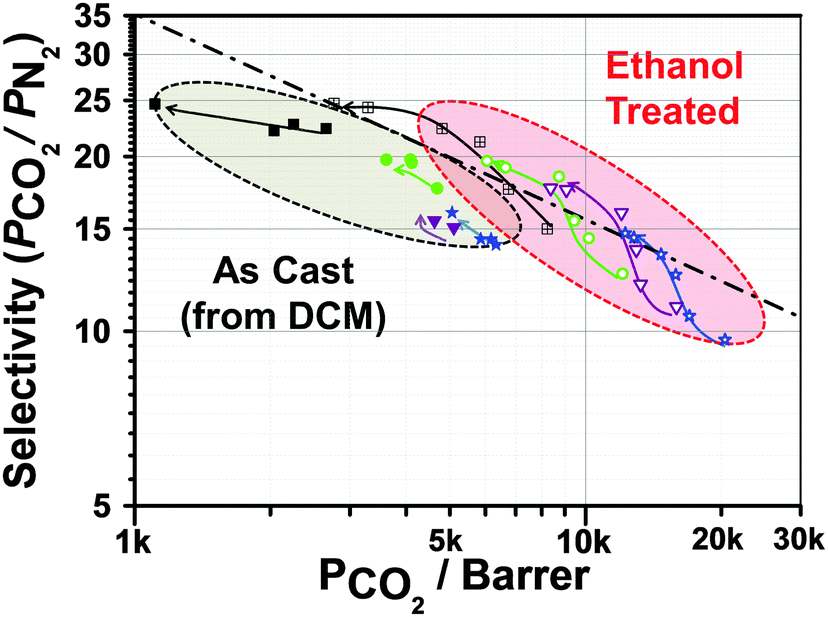 | ||
| Fig. 4 The single gas CO2 permeability and ideal CO2/N2 selectivity of PIM-1 based MMMs plotted against Robeson's 2008 upper bound. The filled squares and squares with the cross represent pure PIM-1 films. The filled shapes represent as cast, the open symbols the analogous films after ethanol treatment. The circles are for a filler content of 5.7 wt%, blue triangles for 16.7 wt% and green triangles for 21.3 wt% (As-cast data: 0 → 45 → 60 → 150 days; ethanol treated data: 0 → 15 → 30 → 45 → 90 → 150 days). Arrows indicate aging direction. | ||
In summary, formation of MMMs from the glassy polymer, PIM-1 and a nanoporous, nanoparticulate HCP, has been described. The integration of an HCP filler in PIM-1 not only leads to higher permeability but also to a significant arrest in polymer aging and permeability loss. We speculate that since fillers are crosslinked polymers the pore diameters of HCP polystyrene are rather more rigid and hence do not collapse due to aging. Since permeability of MMMs are averaged out over the polymeric and dispersed phases, the overall results regards to aging are improved.
Integration of other organic filler such as PAF-1 or CC3 can increase permeability with regards to CO2 up to 320% (see Table 1), while PIM-1/PAF-1 membrane loses only 7% permeability over 240 days. However, these HCP fillers are likely to be more cost effective than other examples in the literature. Moreover, the use of the emulsion polymerization to produce the HCP leads to highly uniform particles that are readily cast from solution to give membranes with excellent filler homogeneity. We believe that this represents a useful design strategy for cost-effective and scalable MMMs for gas separations.
We thank the EPSRC for funding (EP/M001342/1). D. A. thanks the EPSRC for a fellowship (EP/L021978/1).
Notes and references
- J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed.
- Z. R. Herm, E. D. Bloch and J. R. Long, Chem. Mater., 2014, 26, 323–338 CrossRef CAS.
- L. Chen, P. S. Reiss, S. Y. Chong, D. Holden, K. E. Jelfs, T. Hasell, M. A. Little, A. Kewley, M. E. Briggs, A. Stephenson, K. M. Thomas, J. A. Armstrong, J. Bell, J. Busto, R. Noel, J. Liu, D. M. Strachan, P. K. Thallapally and A. I. Cooper, Nat. Mater., 2014, 13, 954–960 CrossRef CAS PubMed.
- D. Banerjee, A. J. Cairns, J. Liu, R. K. Motkuri, S. K. Nune, C. A. Fernandez, R. Krishna, D. M. Strachan and P. K. Thallapally, Acc. Chem. Res., 2015, 48, 211–219 CrossRef CAS PubMed.
- P. Nugent, Y. Belmabkhout, S. D. Burd, A. J. Cairns, R. Luebke, K. Forrest, T. Pham, S. Ma, B. Space, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, Nature, 2013, 495, 80–84 CrossRef CAS PubMed.
- R. W. Baker, Ind. Eng. Chem. Res., 2002, 41, 1393–1411 CrossRef CAS.
- G. Dong, H. Li and V. Chen, J. Mater. Chem. A, 2013, 1, 4610–4630 CAS.
- L. M. Robeson, J. Membr. Sci., 2008, 320, 390–400 CrossRef CAS.
- P. M. Budd, N. B. McKeown and D. Fritsch, Macromol. Symp., 2006, 245–246, 403–405 CrossRef.
- T. Masuda, E. Isobe, T. Higashimura and K. Takada, J. Am. Chem. Soc., 1983, 105, 7473–7474 CrossRef CAS.
- N. Du, H. B. Park, G. P. Robertson, M. M. Dal-Cin, T. Visser, L. Scoles and M. D. Guiver, Nat. Mater., 2011, 10, 372–375 CrossRef CAS PubMed.
- S. Kim and Y. M. Lee, Prog. Polym. Sci., 2015, 43, 1–32 CrossRef CAS.
- J. Kurchan, Nature, 2005, 433, 222–225 CrossRef CAS PubMed.
- L. Shao, J. Samseth and M.-B. Hägg, Plasma Processes Polym., 2007, 4, 823–831 CrossRef CAS.
- S. D. Kelman, B. W. Rowe, C. W. Bielawski, S. J. Pas, A. J. Hill, D. R. Paul and B. D. Freeman, J. Membr. Sci., 2008, 320, 123–134 CrossRef CAS.
- Q. Song, S. Cao, R. H. Pritchard, B. Ghalei, S. A. Al-Muhtaseb, E. M. Terentjev, A. K. Cheetham and E. Sivaniah, Nat. Commun., 2014, 5, 4813 CrossRef PubMed.
- F. Y. Li, Y. Xiao, T.-S. Chung and S. Kawi, Macromolecules, 2012, 45, 1427–1437 CrossRef CAS.
- B. Seoane, J. Coronas, I. Gascon, M. E. Benavides, O. Karvan, J. Caro, F. Kapteijn and J. Gascon, Chem. Soc. Rev., 2015, 44, 2421–2454 RSC.
- A. F. Bushell, P. M. Budd, M. P. Attfield, J. T. A. Jones, T. Hasell, A. I. Cooper, P. Bernardo, F. Bazzarelli, G. Clarizia and J. C. Jansen, Angew. Chem., Int. Ed., 2013, 52, 1253–1256 CrossRef CAS PubMed.
- J. Hradil, P. Sysel, L. Brožová, J. Kovářová and J. Kotek, React. Funct. Polym., 2007, 67, 432–441 CrossRef CAS.
- C. H. Lau, K. Konstas, C. M. Doherty, S. Kanehashi, B. Ozcelik, S. E. Kentish, A. J. Hill and M. R. Hill, Chem. Mater., 2015, 27, 4756–4762 CrossRef CAS.
- C. H. Lau, P. T. Nguyen, M. R. Hill, A. W. Thornton, K. Konstas, C. M. Doherty, R. J. Mulder, L. Bourgeois, A. C. Y. Liu, D. J. Sprouster, J. P. Sullivan, T. J. Bastow, A. J. Hill, D. L. Gin and R. D. Noble, Angew. Chem., Int. Ed., 2014, 53, 5322–5326 CrossRef CAS PubMed.
- C. H. Lau, K. Konstas, A. W. Thornton, A. C. Y. Liu, S. Mudie, D. F. Kennedy, S. C. Howard, A. J. Hill and M. R. Hill, Angew. Chem., Int. Ed., 2015, 54, 2669–2673 CrossRef CAS PubMed.
- R. T. Woodward, L. A. Stevens, R. Dawson, M. Vijayaraghavan, T. Hasell, I. P. Silverwood, A. V. Ewing, T. Ratvijitvech, J. D. Exley, S. Y. Chong, F. Blanc, D. J. Adams, S. G. Kazarian, C. E. Snape, T. C. Drage and A. I. Cooper, J. Am. Chem. Soc., 2014, 136, 9028–9035 CrossRef CAS PubMed.
- B. Li, R. Gong, W. Wang, X. Huang, W. Zhang, H. Li, C. Hu and B. Tan, Macromolecules, 2011, 44, 2410–2414 CrossRef CAS.
- C. H. Lau, X. Mulet, K. Konstas, C. M. Doherty, M.-A. Sani, F. Separovic, M. R. Hill and C. D. Wood, Angew. Chem., Int. Ed., 2016, 55, 1998–2001 CrossRef CAS PubMed.
- B. Li, X. Huang, L. Liang and B. Tan, J. Mater. Chem., 2010, 20, 7444–7450 RSC.
- P. M. Budd, N. B. McKeown, B. S. Ghanem, K. J. Msayib, D. Fritsch, L. Starannikova, N. Belov, O. Sanfirova, Y. Yampolskii and V. Shantarovich, J. Membr. Sci., 2008, 325, 851–860 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc00261g |
| This journal is © The Royal Society of Chemistry 2016 |