
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantioselective bromocyclization of 2-geranylphenols induced by chiral phosphite–urea bifunctional catalysts†
Yasuhiro
Sawamura
a,
Yoshihiro
Ogura
a,
Hidefumi
Nakatsuji‡
a,
Akira
Sakakura
*b and
Kazuaki
Ishihara
*a
aGraduate School of Engineering, Nagoya University, B2-3(611), Furo-cho, Chikusa, Nagoya 464-8603, Japan. E-mail: ishihara@cc.nagoya-u.ac.jp
bGraduate School of Natural Science and Technology, Okayama University, 3-1-1 Tsushimanaka, Kita-ku, Okayama 700-8530, Japan. E-mail: sakakura@okayama-u.ac.jp
First published on 18th March 2016
Abstract
Chiral phosphite–urea bifunctional catalysts have been developed for the enantioselective bromocyclization of 2-geranylphenols with N-bromophthalimide (NBP) for the first time. The chiral triaryl phosphite moiety activates NBP to generate a bromophosphonium ion. On the other hand, the urea moiety interacts with a hydroxyl group of the substrate through hydrogen bonding interactions. Enantioselectivity is effectively induced through two-point attractive interactions between the catalyst and the substrate.
Optically active bromine-containing natural products isolated from marine organisms have been shown to possess several bioactivities such as anticancer and antiviral activities.1 These natural products are biosynthesized by enantioselective bromocyclization induced by enzymes such as vanadium bromoperoxidase (V-BPO).2 For example, in the biosynthesis of isoaplysin-20, the bromonium ion generated in the active site of V-BPO reacts with the terminal olefin of geranylgeraniol site- and enantioselectively. Subsequent diastereoselective π-cation cyclization gives isoaplysin-20.3 While the diastereoselective bromocyclization of linear polyprenoids has been studied for about 50 years,4,5 there have been few reports on the enantioselective bromocyclization of polyprenoids induced by chiral catalysts.6 In 2010, Snyder and colleagues demonstrated enantioselective bromocyclization with stoichiometric amounts of a Hg(OTf)2–chiral bis(oxazoline) complex.6a In 2013, Braddock and colleagues reported that enantiospecific polyene cyclization was initiated by the formation of an enantiopure bromiranium ion.6b However, these methods require stoichiometric amounts of promoters or multiple reaction steps.
Since 2007, we have also developed nucleophilic phosphorous(III) catalysts bearing protic functional groups, 3 and 4, for the halocyclization of polyprenoids (Scheme 1).7–14 Catalysts 3 and 4 activate N-halosuccinimides (X = I and Br) to generate active halophosphonium salt species in situ (Scheme 2). This activation step proceeds smoothly via a mechanism that involves catching a succinimide anion with protons of the catalysts under equilibrium.15 A halophosphonium salt then reacts with polyprenoids at the terminal olefin of 1 to mainly give a halogenated trans-fused AB-ring product 2. Chiral phosphoramidite 3 gave an iodinated product 2 (X = I) with high enantioselectivity.8a However, a stoichiometric amount of 3 was required to give 2 (X = I) in sufficient yield due to strong acid–base affinity between 3 and succinimide. In contrast, low enantioselectivity was observed in the bromocyclization of 1 using 3 under the same conditions.8a More recently, we succeeded in the highly efficient site- and diastereoselective bromocyclization of 1 with the use of a catalytic amount of achiral triaryl phosphite–urea cooperative catalyst 4 to give a brominated product 2 (X = Br) in excellent yield.8c,d Nevertheless, we still have not achieved a catalytic enantioselective halocyclization of 1.
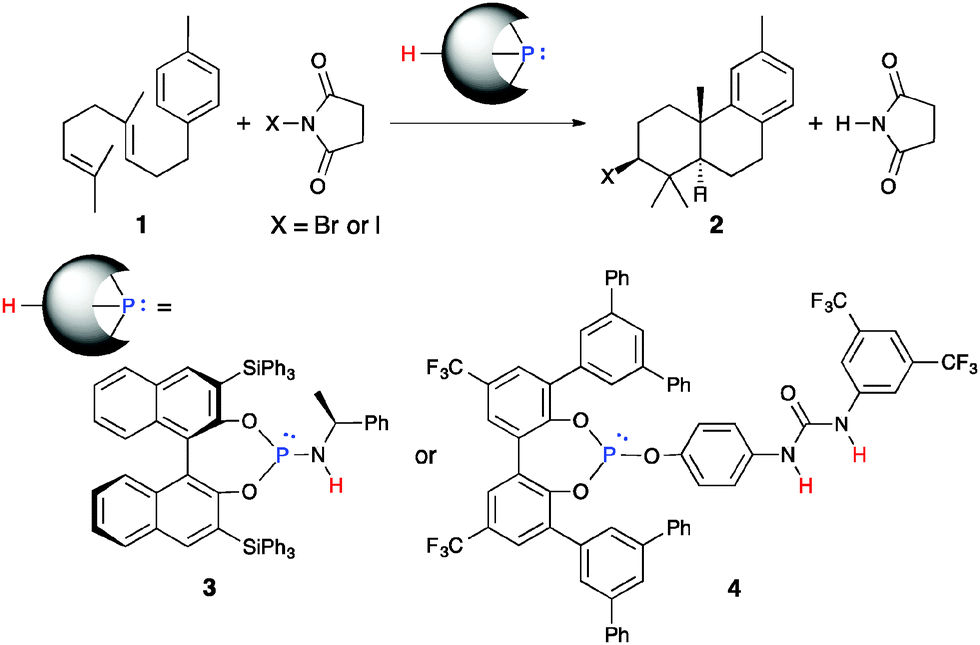 | ||
| Scheme 1 Halocyclization of 1 with nucleophilic phosphorous catalysts (our previous results). | ||
 | ||
| Scheme 2 Proposed mechanism (our previous results). | ||
One reason why enantioselective bromocyclization is difficult is that the three-membered cyclic bromiranium ion rapidly transfers to other olefins.16 In the course of bromiranium ion-olefin transfer, the enantioenriched bromiranium ion is racemized. We envisioned that bromiranium ion-olefin transfer might be suppressed by second non-covalent bonding interaction between a substrate and a catalyst. Here we describe the rational design of chiral phosphite–urea bifunctional catalysts for the enantioselective bromocyclization of 2-geranylphenols.
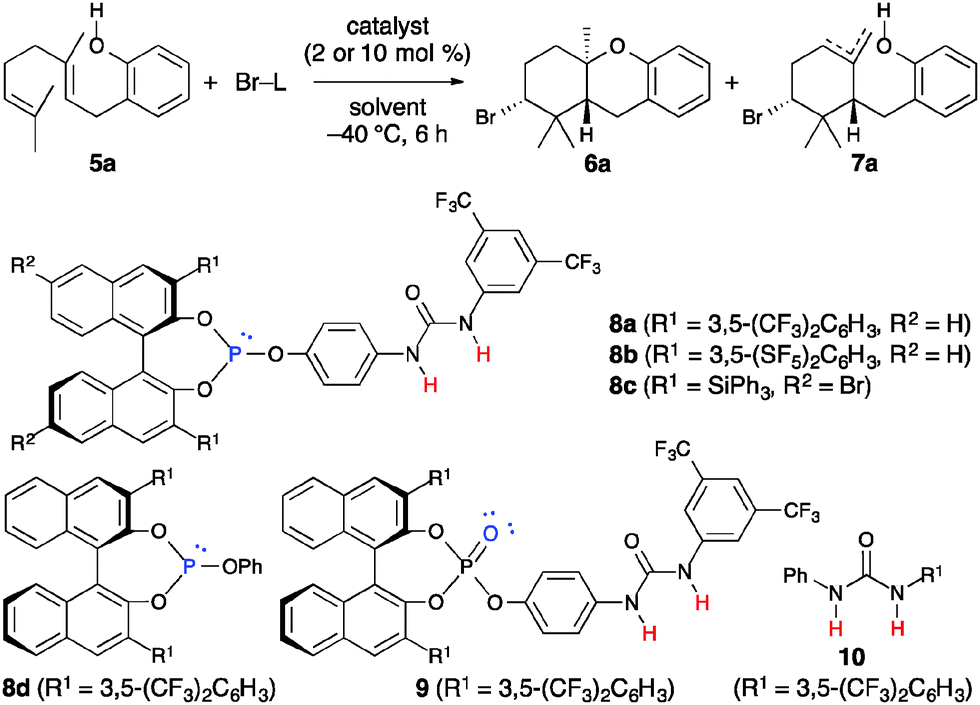
We first examined the bromocyclization of 2-geranylphenol 5a using chiral phosphite–urea bifunctional catalysts 8 (Table 1). The reaction was conducted with 1.1 equivalents of N-bromosuccinimide (NBS) as the brominating reagent in the presence of 10 mol% of 8a in toluene at −40 °C for 6 h. As a result, a trans-fused brominated AB-ring product 6a was obtained in 30% yield with 18% ee together with endo- and exo-isomeric A-ring products 7a in 56% yield with 39% ee (entry 1).17,18 A-ring products 7a could be converted to a diastereomeric mixture of trans- and cis-fused AB-ring products 6a (trans![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :cis = 3:1) by treatment with TfOH, and the enantioselectivity was determined at this stage.19 Interestingly, products 7a were obtained with higher enantioselectivity than 6a. Next, we examined the use of other brominating reagents in place of NBS. Both the reactivity and enantioselectivity were decreased with N-bromoacetamide (NBA) (entry 2). In contrast, the use of 2,4,4,6-tetrabromo-2,5-cyclohexadienone (TBCO) and N-bromophthalimide (NBP) slightly increased the enantioselectivity (entries 3 and 4). Thus, we chose NBP because it was less expensive than TBCO. Next, we examined the solvent effect. The enantioselectivity was decreased with chlorobenzene (entry 5) and the reactivity was decreased with mesitylene (entry 6). The use of 8b gave especially high enantioselectivity for 7a, while the enantioselectivity of 6a was decreased (entry 7). Moreover, when the concentration was lowered to 0.02 M, the enantioselectivity was increased to 65% (entry 8). The enantioselectivity was rather decreased when the reaction was cooled to −60 °C (entry 9). This result suggests that catalysts may aggregate under these reaction conditions. The use of 2 mol% of 8b was also effective, and both 6a and 7a were obtained without any loss of enantioselectivity (entry 10). Catalyst 8c was examined because in our previous studies chiral 3,3′-bis(triphenylsilyl)-1,1′-binaphthol-derived catalysts were effective in inducing high enantioselectivity, such as in the iodo- and protocyclization of polyprenoids8,9 and iodolactonization.11c However, 8c did not induce high enantioselectivity (entry 11). The use of 8a was much more effective than the use of 8d or 8d–10 (entries 12 and 13). Some phosphites [P(III)] are readily oxidized to the corresponding phosphates [P(V)] in the presence of halogenating reagents and moisture or air. Although the reaction was examined using phosphate 9 as a catalyst or without catalysts just in case, 5a was almost recovered (entries 14 and 15). The absolute configuration of 6a and 7a was determined to be (2R,4aR,9aR) by derivatization to a known optically active compound 11a (Scheme 3).19
:cis = 3:1) by treatment with TfOH, and the enantioselectivity was determined at this stage.19 Interestingly, products 7a were obtained with higher enantioselectivity than 6a. Next, we examined the use of other brominating reagents in place of NBS. Both the reactivity and enantioselectivity were decreased with N-bromoacetamide (NBA) (entry 2). In contrast, the use of 2,4,4,6-tetrabromo-2,5-cyclohexadienone (TBCO) and N-bromophthalimide (NBP) slightly increased the enantioselectivity (entries 3 and 4). Thus, we chose NBP because it was less expensive than TBCO. Next, we examined the solvent effect. The enantioselectivity was decreased with chlorobenzene (entry 5) and the reactivity was decreased with mesitylene (entry 6). The use of 8b gave especially high enantioselectivity for 7a, while the enantioselectivity of 6a was decreased (entry 7). Moreover, when the concentration was lowered to 0.02 M, the enantioselectivity was increased to 65% (entry 8). The enantioselectivity was rather decreased when the reaction was cooled to −60 °C (entry 9). This result suggests that catalysts may aggregate under these reaction conditions. The use of 2 mol% of 8b was also effective, and both 6a and 7a were obtained without any loss of enantioselectivity (entry 10). Catalyst 8c was examined because in our previous studies chiral 3,3′-bis(triphenylsilyl)-1,1′-binaphthol-derived catalysts were effective in inducing high enantioselectivity, such as in the iodo- and protocyclization of polyprenoids8,9 and iodolactonization.11c However, 8c did not induce high enantioselectivity (entry 11). The use of 8a was much more effective than the use of 8d or 8d–10 (entries 12 and 13). Some phosphites [P(III)] are readily oxidized to the corresponding phosphates [P(V)] in the presence of halogenating reagents and moisture or air. Although the reaction was examined using phosphate 9 as a catalyst or without catalysts just in case, 5a was almost recovered (entries 14 and 15). The absolute configuration of 6a and 7a was determined to be (2R,4aR,9aR) by derivatization to a known optically active compound 11a (Scheme 3).19
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Cat. | Br–L | Solvent | 6a | 7a | ||
| Yieldb (%) | ee (%) | Yieldb (%) | eec (%) | ||||
| a Unless otherwise noted, the reaction of 5a (0.1 mmol) was conducted with Br–L (1.1 equiv.) in the presence of 8 (10 mol%) in toluene (1 mL) at −40 °C for 6 h. b Determined by 1H NMR analysis using tetrachloroethane as an internal standard. c Determined after treatment with TfOH (4 equiv.) in i-PrNO2 (0.6 mL) at −78 °C for 24 h. d The reaction was conducted in toluene (5 mL). e The reaction was conducted at −60 °C. f 2 mol% of 8b was used for 12 h. g Each 10 mol% of 8d and 10 was used. | |||||||
| 1 | 8a | NBS | PhMe | 30 | 18 | 56 | 39 |
| 2 | 8a | NBA | PhMe | 18 | 13 | 32 | 33 |
| 3 | 8a | TBCO | PhMe | 31 | 21 | 55 | 44 |
| 4 | 8a | NBP | PhMe | 29 | 21 | 57 | 43 |
| 5 | 8a | NBP | PhCl | 35 | 4 | 40 | 19 |
| 6 | 8a | NBP | 1,3,5-Me3C6H3 | 22 | 24 | 47 | 36 |
| 7 | 8b | NBP | PhMe | 25 | 16 | 54 | 51 |
| 8d | 8b | NBP | PhMe | 21 | 19 | 63 | 65 |
| 9d,e | 8b | NBP | PhMe | 21 | 13 | 66 | 61 |
| 10d,f | 8b | NBP | PhMe | 19 | 19 | 60 | 65 |
| 11 | 8c | NBP | PhMe | 21 | −16 | 34 | −21 |
| 12 | 8d | NBP | PhMe | 8 | — | 16 | — |
| 13g | 8d/10 | NBP | PhMe | 29 | — | 26 | — |
| 14 | 9 | NBP | PhMe | 4 | — | 5 | — |
| 15 | — | NBP | PnMe | 0 | — | 0 | — |
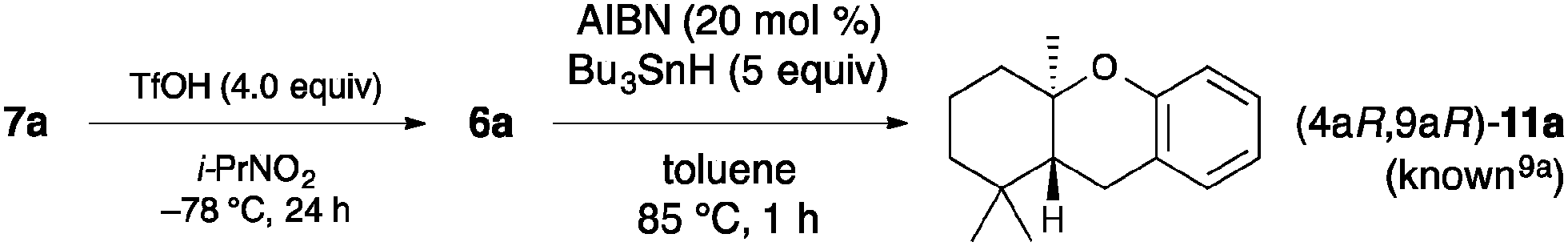 | ||
| Scheme 3 Determination of the absolute configuration of 6a and 7a. | ||
The substrate scope and limitations were investigated under the optimized conditions (Table 2). The results showed that 4- or 5-substituted 2-geranylphenols were suitable as substrates. A-ring products 7 were obtained in good yields with good enantioselectivities (65–71% ee). In contrast, AB-ring products 6 were obtained in low yields with low enantioselectivities (13–29% ee).17 Products 6 and 7 were easily separated by column chromatography on silica gel.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | 5 (X) | 6 | Yieldb (%) | ee (%) | 7 | Yieldb (%) | eec (%) |
| a The reaction of 5 (0.1 mmol) was conducted with NBP (1.1 equiv.) in the presence of 8b (10 mol%) in toluene (5 mL) at −40 °C for 6 h. b Determined by 1H NMR analysis using tetrachloroethane as an internal standard. c Determined after treatment with TfOH (4 equiv.) in i-PrNO2 (0.6 mL) at −78 °C for 24 h. | |||||||
| 1 | 5b (4-CF3) | 6b | 23 | 29 | 7b | 60 | 66 |
| 2 | 5c (4-Br) | 6c | 18 | 21 | 7c | 61 | 71 |
| 3 | 5d (4-OMe) | 6d | 19 | 20 | 7d | 68 | 67 |
| 4 | 5e (4-Me) | 6e | 16 | 19 | 7e | 56 | 67 |
| 5 | 5f (5-Ph) | 6f | 28 | 13 | 7f | 63 | 65 |

Our proposed mechanism is shown in Scheme 4. First, the bromophosphonium ion intermediate 12 should be generated from 8b and NBP in situ. The geometry of 12 is also shown by the Newman projection viewed along the P–Br bond. The terminal alkenyl moiety of substrate 5a reacted with the bromonium ion of 12, which probably minimized steric hindrance for each other. The approach of the bromonium ion to the Si-face of 5a might be disfavored because of steric repulsion between the 3-[3,5-bis(pentafluorosulfanyl)phenyl] group and the dimethylmethylene group of 5a, as shown in 14. Therefore, the Re-face approach via13 might be favored to give (2R)-6a and (2R)-7a enantioselectively.
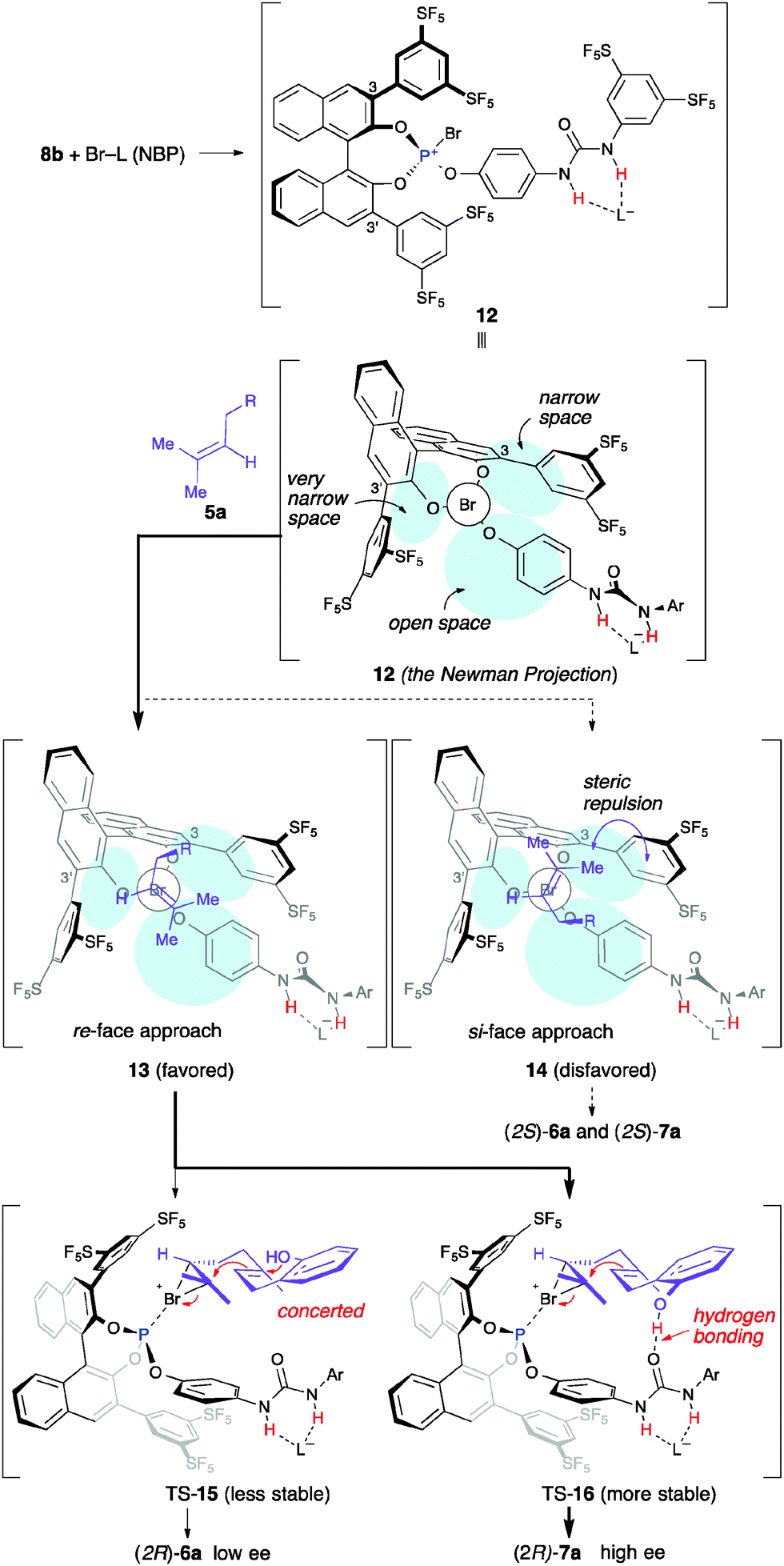 | ||
| Scheme 4 Proposed mechanism for the enantioselective bromocyclization of 5a with NBP catalysed by 8b. | ||
Next, we considered why A-ring products 7a were obtained with higher enantioselectivity than AB-ring product 6a (Table 1 and Scheme 4). The cyclization step to form the A-ring should be different in the reaction pathways to 6a and 7a because both enantioselectivities were not identical. The double-cyclization reaction should concertedly occur via a transition state (TS) 15, since 6a was obtained as only a trans-fused diastereomer. If 6a is formed by a stepwise mechanism, cis-fused product 6a should also be generated as a minor diastereomer. The ee value of 6a was quite low, probably due to rapid racemization of a chiral cyclic bromiranium ion intermediate16 or low enantioface discrimination of the terminal alkenyl moiety of 5a with 8b. In contrast, A-ring products 7a were obtained as major products with good enantioselectivity. The deprotonation of a tertiary carbocation intermediate to give 7a predominantly occurred in place of a second cyclization to give 6a. Hydrogen bonding interactions between the 2-hydroxyl group of 5a and the urea moiety of 8b might dually suppress the second cyclization by decreasing the nucleophilicity of the 2-hydroxy group and controlling its conformation. Furthermore, these interactions might play a role in stabilizing TS-16 to give 7a with high enantioselectivity.20
To ascertain the significance of the hydrogen bonding in the present catalysis, we examined the bromocyclization of geranylbenzene 17 and O-protected substrate 19 under the same conditions (Scheme 5). In both cases, the corresponding brominated products were obtained in good yield with very low enantioselectivity. These results suggest that a hydroxyl group plays a crucial role in asymmetric control.
 | ||
| Scheme 5 Bromocyclization of 17 and 19 with NBP catalysed by 8a. | ||
In conclusion, we designed chiral phosphite–urea bifunctional catalysts for the enantioselective bromocyclization of 2-geranylphenol 5. Catalyst 8b gave A-ring products 7 in good yield with good enantioselectivity. Subsequent treatment of 6 with TfOH gave the trans-fused AB-ring products 6 as major diastereomers. Hydrogen bonding interactions between the urea moiety of 8b and the 2-hydroxyl group of the substrate strongly supported the enantioselective bromocyclization. Further studies on the catalyst to improve enantioselectivity and catalytic activity and investigation of the detailed reaction mechanism are underway.
Financial support was provided in part by JSPS KAKENHI Grant Number 15H05755 and Program for Leading Graduate Schools “IGER program in Green Natural Sciences”, MEXT, Japan. Y. O. is grateful for a JSPS Research Fellowship for Young Scientists.
Notes and references
- For a review, see: B.-G. Wang, J. B. Gloer, N.-Y. Ji and J.-C. Zhao, Chem. Rev., 2013, 113, 3632 CrossRef CAS PubMed.
- For a review, see: A. Butler and M. Sandy, Nature, 2009, 460, 828 Search PubMed.
- (a) S. Yamamura and Y. Terada, Tetrahedron Lett., 1977, 18, 2171 CrossRef; (b) A. Butler and J. N. Carter-Franklin, Nat. Prod. Rep., 2004, 21, 180 RSC.
- For a review, see: S. A. Snyder, D. S. Treitler and A. P. Brucks, Aldrichimica Acta, 2011, 44, 27 CAS.
- C. Recsei, B. Chan and C. S. P. McErlean, J. Org. Chem., 2014, 79, 880 CrossRef CAS PubMed.
- (a) S. A. Snyder, D. S. Treitler and A. Schall, Tetrahedron, 2010, 66, 4796 CrossRef CAS; (b) D. C. Braddock, J. S. Marklew, K. M. Foote and A. J. P. White, Chirality, 2013, 25, 692 CrossRef CAS PubMed.
- For recent reviews on the catalytic asymmetric functionalization of alkenes, see: (a) S. E. Denmark, W. E. Kuester and M. T. Burk, Angew. Chem., Int. Ed., 2012, 51, 10938 CrossRef CAS PubMed; (b) Y. A. Cheng, W. Z. Yu and Y.-Y. Yeung, Org. Biomol. Chem., 2014, 12, 2333 RSC.
- (a) A. Sakakura, A. Ukai and K. Ishihara, Nature, 2007, 445, 900 CrossRef CAS PubMed; (b) A. Sakakura, G. Shomi, A. Ukai and K. Ishihara, Heterocycles, 2011, 82, 249 Search PubMed; (c) Y. Sawamura, H. Nakatsuji, A. Sakakura and K. Ishihara, Chem. Sci., 2013, 4, 4181 RSC; (d) Y. Sawamura, H. Nakatsuji, M. Akakura, A. Sakakura and K. Ishihara, Chirality, 2014, 26, 356 CrossRef CAS PubMed; (e) A. Sakakura and K. Ishihara, Chem. Rec., 2015, 15, 728 CrossRef CAS PubMed.
- For reports on polyene protocyclization with chiral Brønsted bases, see: (a) A. Sakakura, M. Sakuma and K. Ishihara, Org. Lett., 2011, 13, 3130 CrossRef CAS PubMed; (b) M. Sakuma, A. Sakakura and K. Ishihara, Org. Lett., 2013, 15, 2838 CrossRef CAS PubMed.
- For halocyclization of alkenes catalyzed by Lewis bases, see: (a) A. Sakakura and K. Ishihara, Chim. Oggi, 2007, 25, 9 CAS; (b) S. E. Denmark and M. T. Burk, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20655 CrossRef CAS PubMed; (c) Y.-C. Wong, Z. Ke and Y.-Y. Yeung, Org. Lett., 2015, 17, 4944 CrossRef CAS PubMed.
- For recent examples on the catalytic asymmetric iodocyclization of alkenes, see: (a) C. B. Tripathi and S. Mukherjee, Org. Lett., 2014, 16, 3368 CrossRef CAS PubMed; (b) D. W. Tay, G. Y. C. Leung and Y.-Y. Yeung, Angew. Chem., Int. Ed., 2014, 53, 5161 CAS; (c) H. Nakatsuji, Y. Sawamura, A. Sakakura and K. Ishihara, Angew. Chem., Int. Ed., 2014, 53, 6974 CrossRef CAS PubMed; (d) T. Arai, N. Sugiyama, H. Masu, S. Kudo, S. Yabe and M. Yamanaka, Chem. Commun., 2014, 50, 8287 RSC; (e) P. Mizar, A. Burrelli, A. Günther, M. Söftje, U. Farooq and T. Wirth, Chem. – Eur. J., 2014, 20, 13113 CrossRef CAS PubMed; (f) Y. Toda, M. Pink and J. N. Johnston, J. Am. Chem. Soc., 2014, 136, 14734 CrossRef CAS PubMed; (g) Z. Shen, X. Pan, Y. Lai, J. Hu, X. Wan, X. Li, H. Zhang and W. Xie, Chem. Sci., 2015, 6, 6986 RSC; (h) T. Arai, O. Watanabe, S. Yabe and M. Yamanaka, Angew. Chem., Int. Ed., 2015, 54, 12767 CrossRef CAS PubMed.
- For recent examples on the catalytic asymmetric bromocyclization of alkenes, see: (a) Z. Ke, C. K. Tan, F. Chen and Y.-Y. Yeung, J. Am. Chem. Soc., 2014, 136, 5627 CrossRef CAS PubMed; (b) Y. Kawato, A. Kubota, H. Ono, H. Egami and Y. Hamashima, Org. Lett., 2015, 17, 1244 CrossRef CAS PubMed; (c) H. Huang, H. Pan, Y. Cai, M. Liu, H. Tian and Y. Shi, Org. Biomol. Chem., 2015, 13, 3566 RSC; (d) Y. A. Cheng, W. Z. Yu and Y.-Y. Yeung, Angew. Chem., Int. Ed., 2015, 127, 12102 CrossRef PubMed.
- For recent examples on the catalytic asymmetric chlorocyclization of alkenes, see: (a) Q. Yin and S. L. You, Org. Lett., 2014, 16, 2426 CrossRef CAS PubMed; (b) A. Jaganathan and B. Borhan, Org. Lett., 2014, 16, 3616 CrossRef CAS PubMed; (c) C.-L. Zhu, Z.-Y. Tian, Z.-Y. Gu, G.-W. Xing and H. Xu, Chem. Sci., 2015, 6, 3044 RSC.
- For recent examples on the catalytic asymmetric fluorocyclization of alkenes, see: (a) V. Rauniyar, A. D. Lackner, G. L. Hamilton and F. D. Toste, Science, 2011, 334, 1681 CrossRef CAS PubMed; (b) H. Egami, J. Asada, K. Sato, D. Hashizume, Y. Kawato and Y. Hamashima, J. Am. Chem. Soc., 2015, 137, 10132 CrossRef CAS PubMed.
- (a) A. Sakakura, R. Kondo, Y. Matsumura, M. Akakura and K. Ishihara, J. Am. Chem. Soc., 2009, 131, 17762 CrossRef CAS PubMed; (b) Y. Matsumura, T. Suzuki, A. Sakakura and K. Ishihara, Angew. Chem., Int. Ed., 2014, 53, 6131 CrossRef CAS PubMed.
- (a) R. S. Brown, R. W. Nagorski, A. J. Bennet, R. E. D. McClung, G. H. M. Aarts, M. Klobukowski, R. McDonald and B. D. Santarsiero, J. Am. Chem. Soc., 1994, 116, 2448 CrossRef CAS; (b) A. A. Neverov and R. S. Brown, J. Org. Chem., 1996, 61, 962 CrossRef CAS; (c) R. S. Brown, Acc. Chem. Res., 1997, 30, 131 CrossRef CAS; (d) S. E. Denmark, M. T. Burk and A. J. Hoover, J. Am. Chem. Soc., 2010, 132, 1232 CrossRef CAS PubMed.
- The B-ring product (ca. 5–10% yield) and the dibrominated product (ca. 0–5% yield) were obtained as byproducts in all cases.
. - The para-substituted N-[3,5-bis(trifluoromethyl)phenyl]urea moieties of 8a as well as 4 were crucial (Scheme 2): the use of a meta-isomer of 8a reduced not only catalytic activity but also enantioselectivity. See also ref. 8c and 8d.
- The same optical purity was observed for trans- and cis-fused 11a. See the ESI† (p. S16) for details.
- We cannot exclude the possibility of an anion exchange reaction between the phthalimide anion (L−) and 8b or 5a. In this case, good enantioselectivity might be induced through alternative hydrogen bonding interactions between the oxyanion of 5a and the urea moiety of 8b.

Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and characterization data for the starting materials and products. See DOI: 10.1039/c6cc00229c |
| ‡ Present address: School of Science and Technology, Kwansei Gakuin University, Japan. |
| This journal is © The Royal Society of Chemistry 2016 |