
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the outside looking in: redefining the role of analytical chemistry in the biosciences
Dominic J.
Hare
 *ab and
Elizabeth J.
New
*c
*ab and
Elizabeth J.
New
*c
aElemental Bio-imaging Facility, University of Technology Sydney, Broadway, New South Wales, Australia. E-mail: dominic.hare@uts.edu.au; Fax: +61 2 9514 1460; Tel: +61 3 9035 9549
bThe Florey Institute of Neuroscience and Mental Health, The University of Melbourne, Parkville, Victoria, Australia
cSchool of Chemistry, The University of Sydney, New South Wales, Australia. E-mail: elizabeth.new@sydney.edu.au; Fax: +61 2 9351 3329; Tel: +61 2 9351 1993
First published on 22nd February 2016
Abstract
Biomedical research has moved on from the study of the structure of organs, cells and organelles. Today, the key questions that must be addressed to understand the body in health and disease are related to fundamental biochemistry: the distribution and speciation of chemicals, the regulation of chemical reactions, and the control of chemical environments. To see advances in this field, it is essential for analytical chemists to actively engage in this process, from beginning to end. In this Feature Article, we review the progress that has been made towards gaining an understanding of the chemistry of the body, while commenting on the intrinsic disconnect between new innovations in the field of analytical chemistry and practical application within the biosciences. We identify the challenges that prevent chemists from making a greater impact in this field, and highlight key steps for moving forward.
Introduction
In Theodore L. Brown's seminal text, first published in 1977 and now in its 12th edition,1 chemistry was described as the ‘central science’, and nowhere is this more true than the biosciences. Every cellular function, from mitotic division, to energy production, and to eventual programmed cell death, is the result of a chemical reaction, and the same can be said with respect to the initiation of disease. Six of the past ten Nobel Prizes for Chemistry are directly related to the biosciences, with the remainder all having some relevant application to that discipline. One need only look at some of the most significant advances in biology in the past few years, such as the application of the CRISPR/Cas system for gene editing2 or the CLARITY approach visualisation of structure and protein distribution in intact systems,3 to recognise the impact that innovative application of chemistry can have within the biosciences.Importantly, current challenges in biological research are centred on the identification and quantification of chemical analytes: from small molecules (such as signalling molecules, drug metabolites or metal ions) to proteins and subcellular structures. This is therefore an ideal context in which to apply to concepts and techniques of analytical chemistry, whether by adaptation of techniques in other applications, or through the development of bespoke analytical solutions for the biological context. While there are numerous biological analytes of interest, the challenges and opportunities bear many common features. As a result, this Feature Article will focus primarily on metal and metalloprotein analytes by way of example, but the concepts discussed here can be readily applied to any biological analyte.
Broadly speaking, analytical solutions can be divided into (1) technologies – the development and adaptation of spectrometric and spectroscopic instrumentation to gain information about the chemical content of a biological system, and (2) reagents – chemicals that react selectively with species of interest, enabling their specific detection from within the complex environment of the body. In this Feature Article, we focus primarily on various mass spectrometric techniques as examples of technologies, and on selective fluorescent probes as prototypical reagents, designed for use with various technologies such as confocal microscopy and flow cytometry. However, analytical chemistry is multifaceted, and it is more than likely that specialists in alternative techniques, such as vibrational or nuclear magnetic resonance spectroscopy, would present a similar viewpoint from their area of expertise.
Recent years have seen the development of a number of analytical tools spanning the electromagnetic spectrum – from X-ray spectroscopy to NMR spectroscopy. Some techniques, such as mass spectrometry, can provide detail to the level of the atomic nucleus, distinguishing isotopes, while others are able to report on the macromolecular: synchrotron X-ray crystallography, for example, can provide protein structures. Such techniques have been extensively reviewed elsewhere.4,5 Here, we consider the breadth of analytical chemistry's role in the biosciences from the perspective of the analyte (Fig. 1), as opposed to the suite of techniques available. By grouping the analytical techniques in this way, it is clear that each biological question can be answered by multiple techniques. Furthermore, since each technique has inherent advantages and disadvantages, these approaches are therefore complementary to one another. There is therefore much benefit in applying more than one analytical approach to a single investigation, to answer questions that no single technique can achieve on its own. For example, an understanding of the metal homeostasis of a cell requires understanding of its oxidation state and coordination environment, which can be probed by fluorescence microscopy or synchrotron X-ray fluorescence techniques; total metal levels, which can be studied by mass spectrometry methods; and the metalloprotein pool, which can be investigated using immunostaining.6 Similarly, understanding a protein's role cannot be achieved by MS analysis alone; bioinformatics is essential for sequencing the protein from MS data, X-ray crystallography and nuclear magnetic resonance are indispensable tools for determining macromolecular structure, and microscopy plays an important role in determining in vivo distribution.
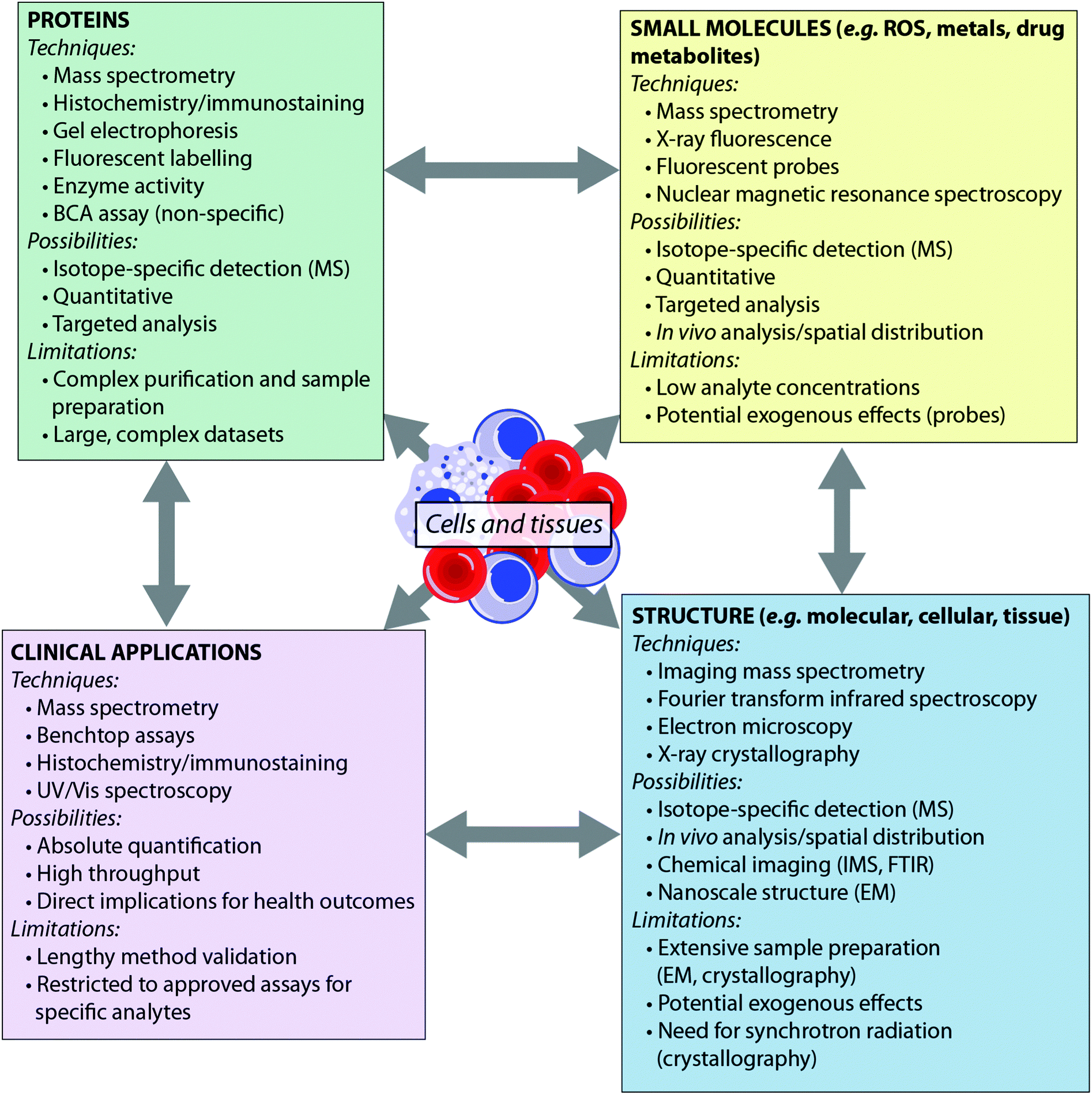 | ||
| Fig. 1 Techniques, applications and limitations of biological analytes in analytical chemistry. | ||
Analytical biochemistry: a dynamic and evolving field
Observing the biochemistry of the cell is itself dependent on our ability to manipulate chemical environments within a biological system to instigate a measurable response. Analytical biochemistry is applied in every biological research paradigm in some way, shape or form; two of the three most cited papers of all time7 report analytical chemistry techniques to study biological systems.8,9 Most mainstream applications are so routine that an understanding of the fundamental principles of these resources is not expected of the typical biologist. This observation is not intended to undermine the ability of members of the biosciences community; rather, it is testament to the successful translation of what would once have been considered complex and labour-intensive protocols into ‘plug-and-play’ devices or simplified ‘recipes’, allowing biomedical researchers to instead dedicate their own time and efforts to delivering essential clinical outcomes.The number of scientists working at the interface of biology and chemistry has exponentially increased in the past few decades. Multidisciplinary science drives almost all innovation in medical research, though there is often a significant delay between the conception of an idea and its translation to providing answers for biological questions. This is perhaps best exemplified in the interaction between chemistry and biology in the development of new drugs. A potential drug must be modelled, synthesised and purified by medicinal chemists, tested for safety and efficacy in animal models by biologists, and translated to the clinic by medical professionals. This whole process is a long, costly, and low-yielding, endeavour. A 2003 study estimated that a single new therapy takes, on average, 15 years and $US800 million to come to market.10 While this can be somewhat attributed to the safeguards in place to ensure adverse effects are identified, which is a process that cannot be rushed, the complexities of the process highlight the heavy dependence on communication between each member of the intellectual production line.
This concept is equally important in the translation of new analytical chemistry approaches to pertinent biological questions, although in this field, where studies are performed on model systems rather than humans, many of the regulatory hurdles can be avoided. While cross-collaboration is more common than ever, there still exists something of a void between those at the cutting-edge of each discipline, which often restricts the immediate uptake of novel advances by those who would most benefit by their application. The tools that have become so integral for studying biochemistry themselves have a long history of development and applications driven by innovation and need. In the context of this Feature Article, we have focused on microscopy and its evolution from a technique used specifically for visualising structure to a broader term describing multiple techniques used for assessing complex chemical interactions at the nanoscale; and mass spectrometry (MS), which recently celebrated its centenary11 and has been a mainstay of the chemical laboratory since its commercialisation in the 1930s.12 As shown in Fig. 2, each technological milestone reached is typically followed by the introduction of new applications relevant to the bioscience, though usually this occurs after some lag time. Microscopy and MS represent only two subdisciplines of analytical chemistry: similar progressive advances can be described for almost every analytical instrument in the modern laboratory, from spectroscopy to chromatography (itself a driver of advances in MS technology). As a general statement, in the modern era, better communication and collaboration between chemists and biologists has significantly reduced the gap between technological innovation and practical biological research.
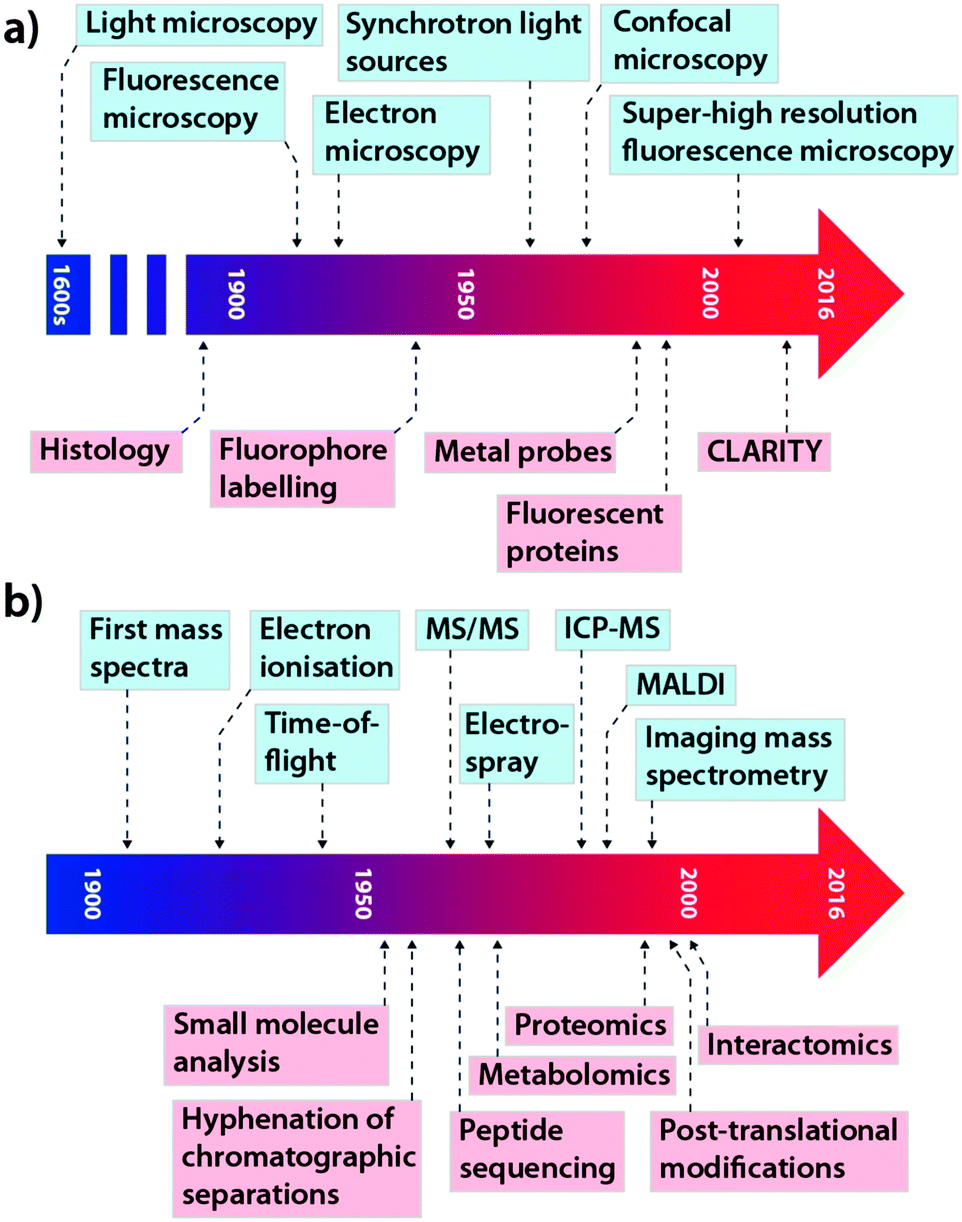 | ||
| Fig. 2 Selected technological (blue) and application (red) milestones in the development of microscopy (a; adapted from ref. 13) and mass spectrometry (b; adapted from ref. 14). | ||
In this Feature Article, we discuss the progress made in further shifting the ‘central’ science towards biology, with an emphasis on our areas of expertise (mass spectrometry (MS), metal analysis and fluorescence sensing). While focusing primarily on our own areas of research interest, we intend this Feature Article to be read with an open mind regarding the wider impacts that analytical chemistry can have on the biological sciences. We outline the steps needed to facilitate the rapid integration of chemistry into both fundamental biochemistry and medical research, and comment on the barriers that still hamper the alignment of two rapidly moving, and sometimes divergent, areas of science.
A potential difference: integrating analytical chemistry into the biosciences
In this Feature Article, our working definition of analytical chemistry is the identification and/or quantification of chemical species. Analytical chemistry played a key role in the derivation of the fundamental laws of chemistry in the time of Lavoisier,15 and has seen enormous advances over the subsequent two centuries. Modern analytical chemistry is sometimes referred to as ‘analytics’, a term designed to encompass not only the measurement and quantification of a sample, but also the increasingly complex data analysis techniques used to identify relationships in complex mixtures, which is particularly pertinent in the biosciences.16 Analytical chemistry techniques continue to be central to numerous fields, from archeology17 to environmental monitoring18 to the pharmaceutical industry,19 and analytical skills remain a key graduate attribute for tertiary chemistry students: British chemistry graduates surveyed 2.5 years after graduation identified analytical chemistry as the most useful of all chemistry knowledge and skills gained in their degree.20 Furthermore, a 2013 survey of over 4000 chemists employed in Europe found that the largest group (17%) worked in analytical chemistry, across research and development, analytical services and quality control sectors.21 However, despite the undisputed importance of analytical chemistry in the workforce, analytical chemistry as an academic discipline has lost favour in recent years, particularly at the most competitive, research intensive universities. For example, at only one of the eight Group of Eight (Go8) universities in Australia is analytical chemistry listed as a research area on the chemistry departmental website. Similarly, of the ten highest-ranked chemistry departments in the UK Research Assessment Exercise of 2014, only two list analytical chemistry as a research strength. This apparent loss of popularity may be due in part to analytical chemists rebranding themselves: whether as environmental chemists, spectroscopists, or, in the case of analytical chemistry for the biosciences, as chemical biologists. In large part, such researchers lack the rigorous training of analytical chemists of the past, and by failing to align themselves with the broader analytical chemistry community, research outputs are not assessed for analytical rigour, nor can they benefit from the wisdom of others engaged in the field. To avoid unhelpful rifts and hierarchies, it is essential that the research world acknowledge the historic and current value of analytical chemistry, that traditional analytical chemists realise the potential of their expertise in a range of sciences, including the biosciences, and that those participating in analytical chemistry research under other guises engage with the analytical community.The first application of analytical chemistry to the biosciences can be traced back to histology, where chemical stains for proteins and cellular features have been used for centuries. After the invention of the light microscope and its first use in the biosciences described by Marcello Malpighi in the 1600s, scientists searched for new and better ways to visualise the components of the cell, and in doing so harnessed the diversity of cell biochemistry to selectively stain unique features, often through simple trial and error. By the turn of the 20th century, histochemical methods were a hotly debated topic amongst its users, with dissensus often coming in the form of unsubtle criticism in the literature. One example is Gōmōri's unrestrained critique, published in 1936,22 of histochemical staining of iron using the Perls and Turnbull protocols favoured by his peers (and both still used frequently today23). Gōmōri's assessment of the methods available included statements such as “[t]herefore I see no reason why the Stoeltzner method,†using a mixture of potassium ferro- and ferricyanide in order to ensure demonstration of both ferric and ferrous compounds, should ever be employed” and “[t]he alleged superiority of the Tirmann-Schmelzer modification of Turnbull's blue method‡is based partly on erroneous theoretical conceptions and partly on the misinterpretation of art[e]facts.” It is noteworthy that even relatively recent studies using this stain still are somewhat unclear as to the specificity of this approach to iron staining,24 particularly with regard to its ability to differentiate iron oxidation states.
This example of the debate that surrounded early microchemical analysis of the cell could be considered a prelude to the difficulties faced by contemporary analytical chemists in applying new techniques, as more is being asked than simply information about the structure and appearance of the cell. In the post-genomic era, snapshots of the chemical makeup of an organelle provides infinitely more information with regard to function, as modulating that function is the goal of those seeking to both understand a biological mechanism or target that function as a therapeutic intervention.
The ‘omic’ revolution (a phrase coined in 2000 by Glen Evans25) has been driven by advances in analytical chemistry, from DNA microarray technology to mass spectrometry. The major ‘omic’ sciences include, but are not limited to, proteomics26,27 (the study of the protein component of the cell), transcriptomics28 (expression profiling, encompassing all RNA molecules), and metabolomics29 (encompassing all breakdown products of metabolic reactions). The latter has also been spun off into numerous sub-omic sciences, recognising that measuring the functional output of the cell requires targeted approaches, such as lipidomics, which focuses on the multifaceted role of long-chain hydrocarbons in the cell and often requires specialised mass spectrometry approaches. Another important addition to the ‘interactome’, a term intended to encompass all the complexities of chemical interactions within the cell30 is the discipline of metallomics,31 which examines the entire metal complement within a cell. Considering that as many a one-third of all proteins contain a metal ion, and one half of all enzymes require a metal to function,32 this relatively new ‘omic’ science (itself integrated into proteomics to reflect the importance of metals and the proteins they interact with33) shares with its peers an unwavering reliance on advancing analytical technology to deliver results. In their 2010 study of two simple microbial organisms with comparatively small proteomes, Cvetkovic et al.34 found that only 50% of the metal-binding species they identified using a fusion of traditional proteomic techniques and chromatographic separations designed to maintain metal–protein bonds could be associated with known metalloproteins. Scaling this up to the human genome, which encodes around 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 proteins (not including the innumerable post-translational modifications that would influence metal binding), a large portion is therefore likely to have interactions with metal ions that are either unknown or poorly characterised.33
000 proteins (not including the innumerable post-translational modifications that would influence metal binding), a large portion is therefore likely to have interactions with metal ions that are either unknown or poorly characterised.33
How analytical chemistry can solve biological problems
‘Omic’ sciences are perhaps the best example of a successful integration of chemistry and biology. There are two divergent lines of enquiry with regard to ‘omic’ sciences and systems biology as a whole: (1) how can analytical chemistry be improved to better answer key biological questions; and (2) are the right questions being asked that take advantage of the tools at our disposal? Desired outcomes are only achievable within the capabilities of the approach used, which can either be a limitation or a significant advantage. For instance, decoding of the human genome relied upon a range of available gene annotation methods, the basis of which was first published in 197735 (referred to a ‘Sanger sequencing’), when the first draft sequence was released in 2001.36 Since, the advent of high-throughput transcript sequencing techniques, such as RNA-Seq,37 have vastly expanded the information from which genome sequencing can provide. In a sense, the field of genomics has stagnated, with papers describing the fully sequenced DNA ‘library’ of an organism no longer considered as impactful as they once were.38 Instead, the arsenal of technologies for both chemical and in silico analysis now available, commonly referred to as ‘next generation sequencing’,39 has dramatically changed the expectations biologists have from analytical approaches, where biochemical variation within a taxa has become of more importance. Loman et al.40 described the current suite of available techniques and platforms for genome and transcriptome sequencing as “an embarrassment of choice”, noting that the rate of improvements in sequencing technology is outpacing that of Moore's law. Without a doubt, biologists focusing on the genetic component of the cell have driven incredibly fast development and application of new analytical technology by asking questions that require innovative answers from analytical chemists. It is unquestionable that the pace of these advances was assisted by the heavy financial support the Human Genome Project received, ($US3.8 billion by its completion in 2003, followed by an additional $US8.5 billion in related research support), which was estimated to have returned $US65 for every dollar spent.41 These (highly controversial) figures only refer to investment by the US government; the total financial support given to the international consortium working on the Human Genome project added even more capital to help drive analytical development.For the fields that have grown from the Human Genome Project, the lack of such a tangible goal as decoding the blueprint for human life means innovation is not driven by the same degree of financial support, and thus technological advances are less frequent. This is not to say that older technology is obsolete or no longer has a place; in several systems biology disciplines this is far from the case. In 2002, Thierry Rabilloud described the place of two-dimensional gel electrophoresis in proteomics as “old fashioned, but it still climbs up the mountains”,42 predicting that 2D gels will “blossom” again in the future. He was correct, with 2D gel electrophoresis still routinely used, though most advances in the method have come from improvements in digital image analysis and amplification of detectable protein signals (referred to in the discipline as differential imaging gel electrophoresis, or DIGE),43 rather than improvements to the physical separation of the proteins in a complex mixture by electrophoresis. However, rapid advances in liquid chromatography (LC) over the past decade, driven by the miniaturisation of separation devices and constantly improving integration of these innovations with MS technology, have begun to overshadow gel electrophoresis as the mainstay of proteomic technique. Microchip assemblies, made possible through the advent of advanced nanofabrication technology (which itself, no doubt, lagged behind in terms of its uptake by the analytical chemistry community), permit not only high resolution separation of proteins, peptides and metabolites using minimal sample volumes, but also allow for on-chip protein enrichment and desalting in a high-throughput and often customisable format.44 These microchips, manufactured at a high quality standard, are very reproducible, and interface well with many MS systems.
Thus, it begs the question, why haven't chip and other LC-based separation approaches completely usurped 2D gels in the proteomics hierarchy more than they have? Unger et al.45 considered the contribution that multi-dimensional LC may have as an alternative to 2D gels in 2000, concluding that development “will probably last a decade until high-performance and rugged systems will be on the market for the comprehensive on-line approach.” This has been the case, though rather than supplanting 2D gel electrophoresis, LC-MS analysis has instead complemented it. Workflows employing both techniques are designed to capitalise on the advantages of both approaches, reducing the chemical complexity of the sample and improving sensitivity for the target analyte.46
An important consideration in the academic research world is, however, cost. A fully stocked proteomics (or any other ‘omic’ lab relying on MS technology) is a capital investment that in on par with the highest expenses in medical research, such as magnetic resonance imaging infrastructure. In the highly competitive academic world, proteomics laboratories are usually built in a modular fashion, as opposed to a major initial investment, with new technology added as funds become available. Alternately, universities and research institutes may invest in centralised facilities, which are ideal for providing a service based on standardised protocols, but rarely afford time for method development. Thus, outside of private industry, which can allow the outlay but is reluctant to share new, potentially lucrative developments in the public space, the application of new innovation is often stymied by a lack of investment in future technologies, which has perhaps kept older technologies like 2D gel electrophoresis very much alive. Analytical manufacturers are recognising this limitation, from both an altruistic and commercial perspective: research breeds innovation, and much work at the coalface of medical research is being undertaken at publicly and philanthropically funded institutions.
Consider the advances in MS over the last decade, and the fact they can be attributed to the tireless work of physicists and engineers, and not the biologists who will ultimately use the technology. We examined the yearly publication output for three types of mass spectrometry tools used in biological research: the inductively coupled plasma-mass spectrometer (ICP-MS; launched commercially in 198347 and used for highly sensitive analysis of metals and bioelements47); the matrix-assisted laser desorption-time of flight-mass spectrometer (MALDI-TOF-MS; launched in 199048 and for which John B. Fenn and Koichi Tanaka were awarded the Nobel Prize for Chemistry in 2002 “for the development of methods for identification and structure analyses of biological macromolecules”); and the Orbitrap MS (200549), a super-high resolution Fourier transform mass analyser critical for contemporary proteomics due to its unparalleled mass accuracy (Fig. 3). Clearly, there is a significant lag in output following the commercialisation of a new technology, and this can be attributed to multiple factors. For ICP-MS, a system initially designed for the geological and environmental sciences, applications within biology were not realised until the mid-2000s, and thus the technique has seen a steady growth since. MALDI-TOF-MS, an immensely popular technology during the chrysalis of proteomics as a discipline in its own right, showed near-exponential growth until the mid-2000s, where it has since plateaued, and the recent downturn in 2015 may be indicative of the technology being superseded by improved technological rivals, such as electrospray ionisation MS interfaces. The Orbitrap design, proprietary to Thermo-Fisher Scientific, is an interesting case study in itself, in that in-house published works prior to its commercial release helped prepare the proteomics community for the introduction of what would become a highly sought-after and significant capital investment. This example is an encouraging sign, where the manufacture of a MS system clearly targeted to life scientists was marketed directly to the end user, and has helped the measureable output of this technology grow steadily over the following decade. These three technologies are just a few examples of high-end technology either finding a niche in the life science space, or, as is the case with ICP-MS, undergoing something of a renaissance once discovered by biologists, which has in turn driven the development of new variants specifically aimed at the life and medical research communities (such as the triple quadrupole ICP-MS, designed in part to meet the needs of high accuracy phosphorus analysis for studying post-translational modifications in proteomics,50 whilst still having a foot firmly planted in the traditional ICP-MS marketplace51). Even established biological MS approaches themselves are able to find new life through diverse applications; one need look no further than the explosion in interest of imaging MS using MALDI, pioneered by Richard Caprioli in the late 1990s52,53 and now being realised as a valuable clinical tool, particularly in cancer research.54
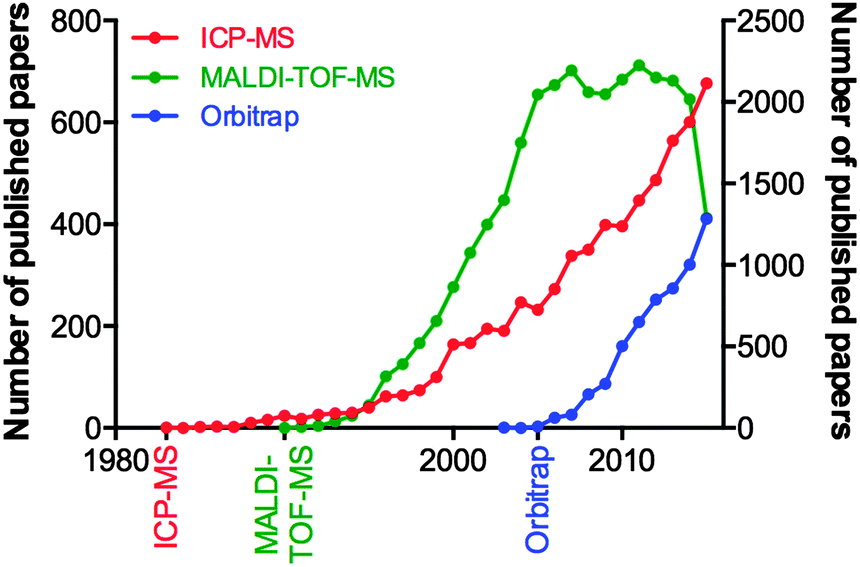 | ||
| Fig. 3 Publication output in biological sciences for ICP-MS, MALDI-TOF-MS and Orbitrap mass spectrometers from initial commercialisation (marked on the x-axis) to 2015 (source: PubMed, accessed December 2015). ICP-MS and Orbitrap designs are shown on the left y-axis; MALDI-TOF-MS on the right. | ||
Clearly, the most commonly asked questions of analytical chemistry with respect to MS by the biosciences are: (1) how much can I detect; (2) how fast can I do it; and (3) what flexibility do I have with the type of analytes I'm interested in? As discussed above, the chemical makeup of a cell has become the new frontier for biologists, and sensitivity, throughput and versatility drives both need and development of new technology. In their colloquium on the history and future directions of MS technology, engineers Maher et al.55 identified rapid analysis as being the main direction MS research should be headed in. Beyond this, and a question that is somewhat outside the scope of this article yet still should be acknowledged is: what on Earth do I do with all this data? Much like communication between analytical chemists and biologists is key to delivering useful outcomes relative to health research, giving biologists the power to sort through data without suffering from the proverbial search for a ‘needle in a stack of needles’ is reliant on collaboration with software developers and bioinformaticians. For a general overview and comprehensive list of relevant reading, we direct the reader to the excellent review by Douglas Kell.56
New questions being asked by biologists are also giving a new lease on life to well established analytical techniques. Gas chromatography (GC), which is still widely used in high-throughput settings around the world, is also finding new applications for small molecule analysis, metabolomics and within the pharmaceuticals industry.57 The excellent resolution, peak capacity, sensitivity and potential for orthogonal chromatography coupled to highly sensitive MS makes GC an attractive alternative to LC for certain analytes. One particular example of interest is the application of GC-MS to lipidomics; fatty acids are well suited to GC separation, can be derivatised en masse, and have achievable detection limits in the femtogram range.58 As a hyphenated technique, GC also benefits from the continuous improvements made to MS technology, and the number and size of GC-MS reference libraries (e.g. the National Institute of Standards and Technology NIST 14 catalogue of nearly 250000 unique compounds) is constantly growing.59
Not all as it seems: the importance of correct data interpretation
Analytical chemistry has, without a doubt, contributed enormously to studies of the biosciences. There is a potential, however, for overinterpretation of experimental results, if investigators fail to recognise the limitations of a technique. Importantly, it is essential that in selecting an appropriate analytical technique to use, researchers identify the specificity of the method, and interpret data collected accordingly.The specificity of an analytical technique refers to its distinction between chemical species, whether different chemical elements or different speciations of the same element. Mass spectrometry methods, for example, can pinpoint exactly a single isotope, but cannot report on the oxidation state or speciation of that isotope. On the other hand, fluorescent probes can be selective for individual oxidation states of metal ions,60,61 or metal coordination environments,62,63 but in some cases distinction of different analytes of similar properties is less straightforward.64,65 Persistent incorrect interpretation of specificity can severely hamper research in a field. For example, in the field of cisplatin chemotherapeutics, intracellular platinum levels are routinely examined by bulk mass spectrometry techniques, which can distinguish only total platinum levels, without sensitivity to oxidation state and coordination environment. However, such data is often reported as ‘intracellular cisplatin concentration’,66,67 suggesting that cisplatin remains intact within the cell, rather than existing primarily as protein-bound adducts, as is now understood.68
Speciation of chemicals within biological systems also requires appreciation of the reactivity of the chemical in its particular environment. This is true of metal ions, amongst other species, for which the speciation strongly determines cellular function or dysfunction. Not only is the oxidation state of a redox-active metal crucial to appreciating its activity, but the coordinating ligands also play a key role. Metal ions may be tightly bound to proteins, eliciting activity at the protein active site, but having little capacity to rapidly move within the cell, or they may be loosely bound to cytoplasmic small molecules or proteins. The latter pool, commonly termed the labile or bioavailable pool, will be much more readily accessible for exchange, and it is perturbations in the levels of the labile metal pool that are believed to be involved in disease.6 In different circumstances, biological research questions may require study of the protein bound metal pool, or the labile metal pool, or total cellular metal, or some combination of the three. It is highly likely, for example, that the total metal level remains constant in response to a stimulus, while the protein-bound and labile pools vary inversely, and measurement of the total metal pool alone would fail to identify any changes. It is essential, therefore, to recognise which analytical techniques can address each pool (for example, laser ablation (LA)-ICP-MS or solution nebulisation ICP-MS for the total metal pool; western blotting or immunostaining for protein-bound metal; imaging using selective probes for the labile metal pool), and where appropriate, to use multiple techniques in parallel to best build a picture of metal distribution throughout the cell (see Fig. 4).
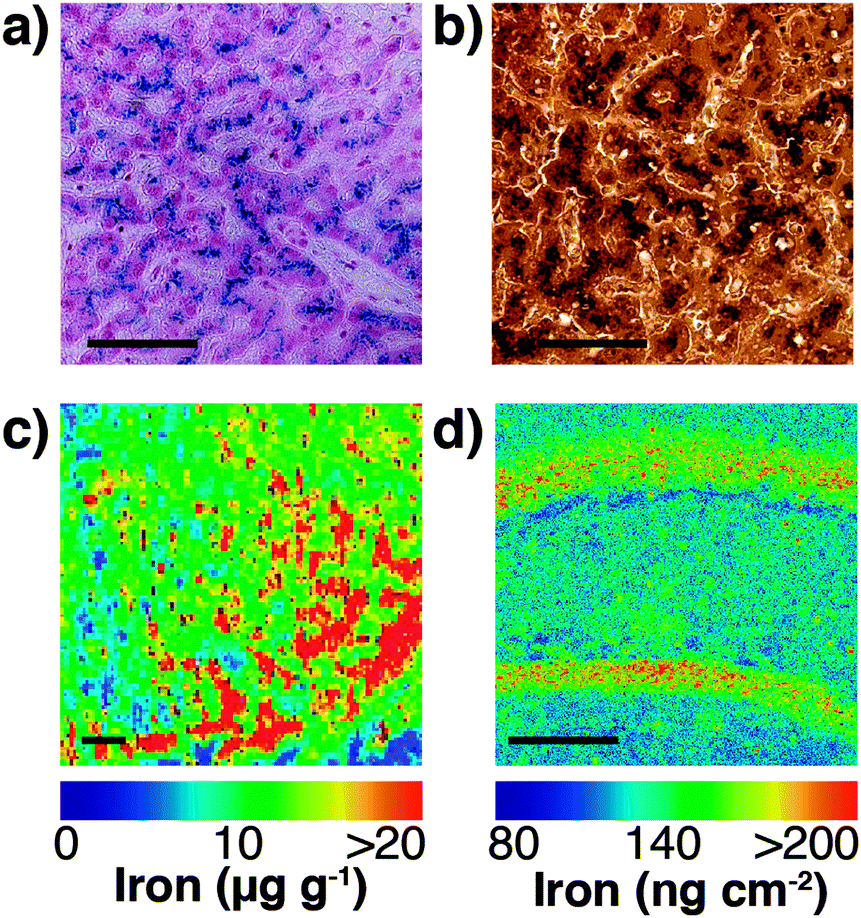 | ||
| Fig. 4 (a) Traditional Perls stain of liver showing non-heme iron deposits in the characteristic blue hue of the Prussian blue pigment (Fe4[Fe(CN)6]3·xH2O). (b) 3,3′-Diaminobenzidine (DAB) and cobalt-enhanced Perls stain (shown here in liver tissue with neutral red counterstain) is reported to be more sensitive than Perls staining alone.69,70 Neither method is quantitative, though both are specific to non-heme iron, such as ferritin-bound Fe3+. (c) LA-ICP-MS mapping of iron in the mouse substantia nigra pars reticulata (adapted from Hare et al.;71 copyright 2014, Royal Society of Chemistry) is both more sensitive and quantitative (with matrix-matched external calibration standards72,73), though image resolution is limited to the μm-scale (pixel size depicted here is 25 μm2) and is sensitive to all chemical states of iron. (d) Synchrotron-based X-ray fluorescence microscopy of the mouse brain here shows marked iron deposition within the molecular cell layer of the dentate gyrus, depicted here quantified against a thin standard film of iron and at a spatial resolution of 4 μm2. Like LA-ICP-MS, XFM produces images of total iron distribution. All scale bars = 50 μm. | ||
In addition to studying the speciation of analytes within the cell, it is also essential to consider their localisation. Although organelle structure is rarely the feature du jour of contemporary cell biology, visualisation of organelle chemistry most certainly is. Advanced fluorophores and super-resolution microscopy have made assessing the chemical composition of the smallest cellular features near routine. Biologists often search for colocalisation of two fluorescent probes as evidence of biological interplay between two variables, and while this may be indicative of some kind of relationship between the analytes, image resolution is not sufficient to definitively interpret apparent overlapping structures as being colocalised.74
Localisation of a molecule or analyte within a cellular structure is still of great importance, though thorough and rigorous interpretation of the data is required, including consideration of all possible confounding factors. It is also essential to consider the chemistry of the two (or more) analytes, when colocalisation is intended to indicate a chemical relationship. Take the example of imaging distinct pools of metals in biological cells.6 Metals may be ubiquitous throughout the body, though, as demonstrated by the emergence of metallomics and metalloproteomics, the function of a metal is driven by both its oxidation state and the biological ligands to which it is bound. Imaging techniques, such as LA-ICP-MS75,76 provides an extremely high level of sensitivity and specificity, yet can only report on the total metal content within a defined region (typically at the μm scale). In a recent paper by Hare et al.,71 LA-ICP-MS was used to examine the relationship between iron and dopamine, where the enzyme tyrosine hydroxylase (the rate limiting step in dopamine synthesis) was tagged with a gold nanoparticle labelled antibody. While this study provided important data regarding the increased oxidative load imparted on dopaminergic neurons in Parkinson's disease by these two chemicals, it can only provide a partial picture of the chemistry driving oxidative stress within these vulnerable cells. Dopamine breakdown facilitated by iron results in the formation of numerous neurotoxic metabolites,78 though this mechanism relies upon the availability of ferrous (Fe2+) ions, which are typically in (deliberate) short supply within the cytoplasm due to its potent redox activity. Therefore, conclusions indicative of a confirmed relationship between iron and dopamine within these neurons cannot be achieved through colocalisation studies alone. Excess iron may be rendered redox-inactive by ferritin,79 or the dopamine itself may be inaccessible to iron through intracellular safe storage, such as in vessicles.80 Only through further intervention, in this case the addition of a potent neurotoxin known to elevate iron levels81 and measuring response can we obtain additional evidence of correlation between these two distinct chemicals, though this is far from providing direct evidence of causation.
This highlights a significant shortcoming in colocalisation analysis and the search for correlation: that of sensitivity. It is plausible that significant colocalisation, and thereby indicators of correlation are not detectable due to the extremely low abundance of analytes that belies their biological significance. Detection of the fluorophore, or even a low abundance biometal, is dependent on the sensitivity of the analytical method being used. This may be related to the fluorescent yield of the fluorophore employed, the sensitivity of the detection technique, or a combination of both factors. As we will discuss in more detail, the development of next generation fluorescent compounds for super-resolution microscopy is a fluid and dynamic space, yet the uptake of these new sensors in the biosciences, like the aforementioned example with mass spectrometry, is dependent on the users, not developers. A further significant limitation in colocalisation analysis and the search for correlation is that of sensitivity. It is plausible that significant colocalisation, and thereby indicators of correlation are not detectable due to the extremely low abundance of analytes that belies their biological significance. This was illustrated in our recent investigation of the colocalisation of biometals in Caenorhabditis elegans, visualised using synchrotron X-ray fluorescence microscopy (XFM; Fig. 5).77 Since strontium is known to substitute for calcium in biological systems,82 we might have expected complete correlation of the two metals, but this was only observed in the anterior gut region. The reason for this disparity was one of sensitivity; XFM, though highly sensitive to calcium at biological concentrations, was only able to discern strontium distribution in areas of high abundance of this element.
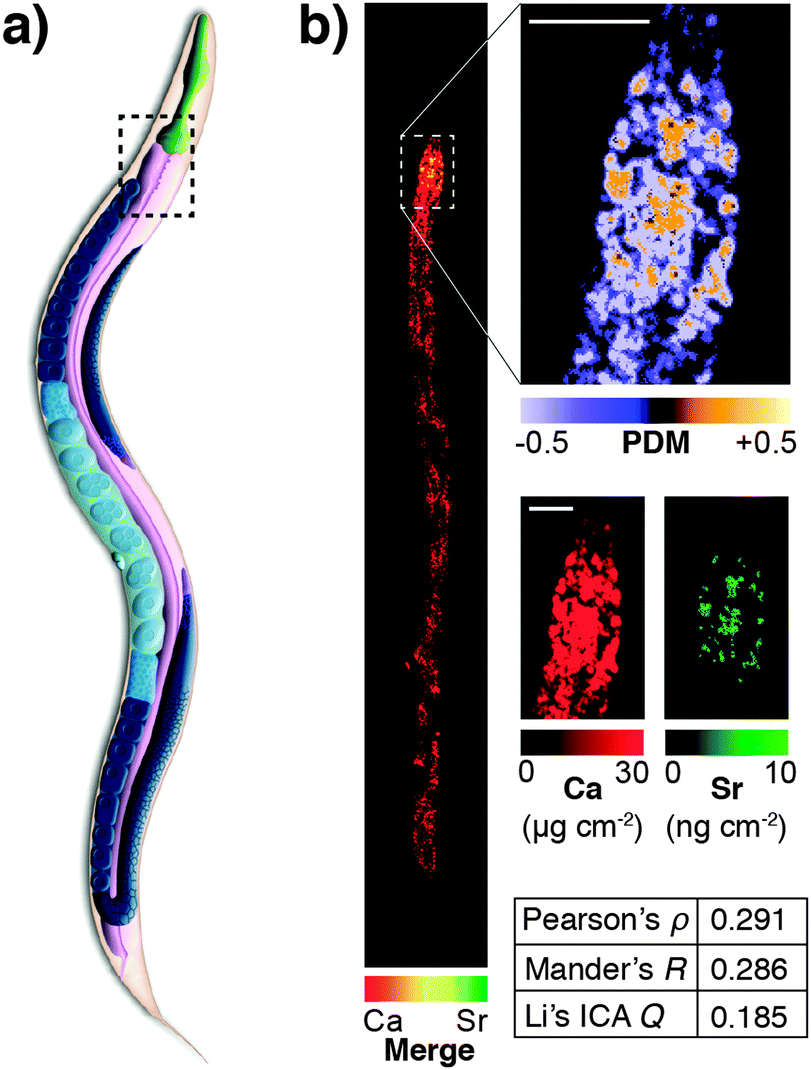 | ||
| Fig. 5 (a) Simplified anatomical structure of Caenorhabditis elegans, with the anterior intestine marked with dashed black box. (b) XFM mapping and Li's intensity correlation analysis on quantitative calcium and strontium maps shows colocalisation and correlation (as according to Pearson's rho, Mander's R and Li's ICQ measures) in the anterior intestine, though a lack of sensitivity for strontium, which is over 1000 times less abundant than calcium, precludes any correlation analysis outside this area of high concentration. Figure adapted from Hare et al.77 (Copyright 2016, Royal Society of Chemistry). | ||
Interpretation of ‘big data’ that is now a common output of analytical chemistry, particularly in the ‘omic’ sciences, has also brought about a need for input from biostatisticians and bioinformatics experts. Chemometrics is not a new concept in analytical chemistry, with multivariate analysis and artificial neural networks for machine learning approaches used with great success to improve analytical separation and classification of complex mixtures.83 In the biosciences, this approach has become integral in the sub-discipline of metabonomics, which differs from metabolomics in that it examines the multivariate effects on a system rather than focusing on specific metabolites.84 An overview of chemometric applications to large datasets from metabonomics, in addition to genomics and proteomics studies can be found in the review by Eriksson et al.85
Past, present and future: tools for analytical chemistry in the biosciences
There is no shortage of chemical analysis techniques that are available to the modern biologist. In the field of metal imaging alone, a vast array of technologies, from mass spectrometry to emission spectroscopy and fluorescent sensors, a biologist has the freedom to pick and choose the appropriate technique that is suited to the application he or she is using.86 The sole limitation is accessibility, from either a practical perspective (i.e. access to synchrotron-based radiation is restricted to around 40 facilities worldwide); or a lack of ability to apply what can be quite complex experimental protocols. Interface scientists, who straddle both the development of analytical techniques and their application to the biosciences are a valuable commodity, as is embedding these scientists within the biological research laboratories who will benefit most from their expertise.The primary role of the analytical chemist in this process, then, is the development of new tools, or the application of existing tools in new ways, to drive forward biological research. In order to achieve greatest impact, it is essential to consider the most valuable avenues for future research. This consideration begs two questions: (1) is effort being exerted towards the most pertinent biological questions? (2) Are current tools being utilised to best effect in biological research?
In addressing the former, it is clear that much research effort is directed towards analytes that are easy to sense, rather than being the most significant. According to Web of Science, in 2015 alone there were over 110 publications describing new fluorescent sensors for inorganic mercury ions, 35 of which cite the endpoint application as measurement of exogenously added mercury in cultured cells. It is unsurprising, however, that mercury sensors are rarely used in detailed biological investigations, as endogenous total mercury levels in cells are orders of magnitude below toxic concentrations.87
On the other hand, there is great value to be gained when analytical tools are developed or modified in response to scientific need. Amongst many other examples is the recently commissioned Bionanoprobe at the Advanced Photon Source. Synchrotron XFM has proved to be an invaluable analytical tool, simultaneously providing multiple element maps.88 However, recent studies suggested that results might be compromised by conventional sample preparation, such as fixation, embedding or dehydration.89 As a result, the Bionanoprobe was developed, which enables cryogenic transfer and mounting of samples, to avoid these sample preparation steps, thereby giving exquisite information of trace elements in their native environments.90
Rather than just focusing on the ‘wants’ of the biologist, however, it is important to appreciate that many analytical techniques are rarely pushed to their extreme limits, and thus the capabilities of modern equipment are often underutilised. LA-ICP-MS is a prime example in the biosciences. First proposed as a sample introduction technique for ICP-optical emission spectroscopy in 198391 and then adapted for mass spectrometry detection in 1985,92 LA-ICP-MS quickly developed a strong following in the earth and environmental sciences,93 as it offered a viable alternative to the labour-intensive and often hazardous sample preparation procedures that employed strong acids and high-pressure microwave digestion. The first imaging application of a biological sample was reported in 1994, where strontium and calcium ratios in fish scales were mapped at 30 μm resolution,94 an impressive achievement considering contemporary LA-ICP-MS mapping over 20 years hence is still only practical at the 1 μm level, and even then this requires specially designed ablation chambers that have been developed only during the past few years.95
Three main factors drive development of new analytical equipment: improved sensitivity, ease of use, and reproducibility. Sensitivity can be improved in a number of ways, from the development of specialised analytical methods specific to a target analyte to improvements of analytical hardware and electronics that are responsible for the physical detection of a molecule (in the case of MS) or characteristic emission profile (for fluorophores). Simplifying the process is key to better integrating technology into biological laboratories. For many scientists, understanding the fundamental physics and chemistry that dictate how an analytical system works is often irrelevant to them, beyond the need to perform routine maintenance and troubleshooting. This is not unreasonable, and the ‘black box’ paradigm of analytical instrumentation has forced manufacturers to rethink both instrument front- (that is, the software controlling them) and back-ends (the means by which data is extracted and processed). There will always be room for improvement in this area, though the advent of Electronic Laboratory Notebooks (ELNs) and Laboratory Information Management Systems (LIMS), many of which are open source,96 is encouraging. Proprietary software and data management systems, such as Agilent Technologies' OpenLAB, are also finding a place within high throughput laboratories and will soon permeate into the research space. Remote access (including control of multiple manufacturers' systems) and multiple redundancies for data storage forms a pipeline of information output that is accessible to both users and stakeholders, and by centralising quality control, maintenance requirements, data collection and analysis, and report dissemination, these tools (both open source and commercially produced) undoubtedly represent the future of analytical chemistry with respect to its integration with medical research and clinical sciences. However, reproducibility, where analysis can be performed at the same standard in any equivalent laboratory setting, is probably the most important factor in expediting the uptake of analytical chemistry research by such researchers.
The development of new analytical reagents, such as fluorescent probes, also follows these same lines. After identifying pertinent analytes to target, typical probe design involves tethering of a sensing element (designed to selectively interact with the analyte) to a fluorophore, in such a way that the interaction of analyte with the sensing group modulates fluorescence of the fluorophore. Probe design tends to be centred around standard classes of fluorophores.97 The primary challenge in sensing a new analyte is therefore the identification and validation of sensing elements that exhibit the required selectivity and sensitivity for the particular application. However, photophysical and biological properties of the probe must also be modulated to ensure best effect. A large class of zinc-sensing probes are based around the fluorescein scaffold, exemplified by the first probe of its type, Zinpyr-1 (Fig. 6).98 This class contains many examples of simple modifications that give rise to improved photophysical and/or biological behaviour. In general, zinc sensors are based on the common, well-established sensing element containing pyridyl groups or similar, and key modifications centre around the remainder of the structure. Increased dynamic range gives rise to lower detection limits and therefore greater sensitivity.99 Ratiometric probes, which signal changes in analyte through changes in emission wavelength rather than emission intensity, enable less ambiguous interpretation of data.100 Many biological questions are centred on specific organelles or cell types, so the ability to target probes to specific regions of the cell or the body will be crucial for future probe design. This can be achieved by the incorporation of small molecule targeting groups, the triphenylphosphonium moiety for mitochondrial localisation,101 or by the use of short peptide sequences.103 Finally, a key challenge in probe design is the development of more red-shifted emitters, which require less damaging, lower energy excitation sources, and enable some degree of tissue penetration. This can be achieved by the use of the related xanthene-based fluorophore, rhodamine.102 In this domain, replacement of the xanthene oxygen with a silicon atom gives rise to near-IR emission, successfully utilised in preparation of a Ca2+-selective sensor.104
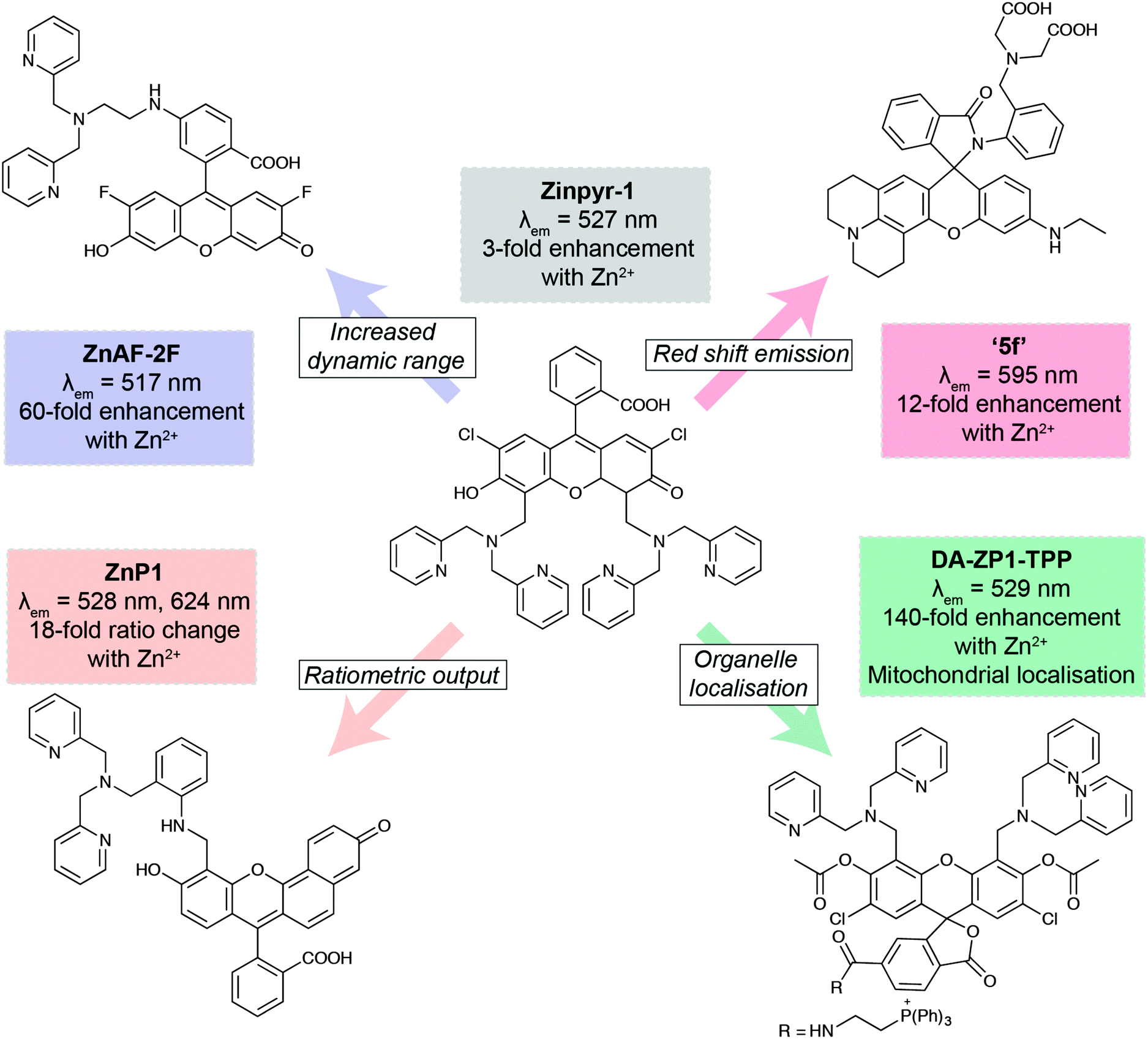 | ||
| Fig. 6 Key developments in xanthene-based fluorescent sensors for Zn2+. Zinpyr-198 has been further developed to improve than range of Zn2+ concentrations it can detect (ZnAF-2F);99 measure Zn2+ flux via ratiometric measurements (ZnP1);100 localise within specific organelles, such as mitochondria (DA-ZP1-TPP);101 and optimise its fluorescence emission peak (5f).102 | ||
When considering the potential application of fluorescent sensors into the clinic, excitation and emission wavelengths are key, with wavelengths of greater than 700 nm required for sufficient tissue penetration.105 The development of new fluorescent scaffolds with such properties is essential to realise this aim. To date, indocyanine green (λem ∼ 810 nm) and methylene blue (λem ∼ 700 nm) are the only two clinically-approved near-IR fluorophores, so there is certainly much work be done in this area.106
Quality control: the key to translation
This question of clinical translation is also pertinent in the domain of mass spectrometry, but the technology it must first overcome the perceived limitations with regard to quantification before this can be fully realised.107 This is not to say that there have not been dramatic strides made in the development of validated analytical protocols for quantitative proteomics,108 and many manufacturers market MS products as being suitable for such a task. Quantitative clinical proteomics must, however, undergo rigorous validation for each individual assay, a process that can be time consuming, expensive and complex. In 2007, Mischak and colleagues109 presented a manifesto outlining all areas where standards should be set for clinical proteomics to become mainstream. Fortunately, global think tanks like the Working Group of the International Federation of Clinical Chemistry and Laboratory Medicine (IFCC) exist to set guidelines for clinicians, such as their recently-released compendium of quality indicators designed to detect pre-analytical errors (that is, extra-laboratory influences impacting sample quality) before a single sample is analysed.110Like any analytical method, validation of an assay intended for use in a clinical setting must follow the defined steps to ensure results and their interpretation are true. Green111 described these steps, to be performed in sequential order, as:
• Establishment of minimum criteria, where the requirements for accuracy and precision are defined. This can be approached on a case-by-case basis, though for diagnostic tests this is of critical importance.
• Demonstration specificity, where the response of the intended analyte is proven to be independent of any other interfering species.
• Demonstration of linearity, where not only are five levels of standards required for quantitative analysis, they must also bracket the expected concentration of the analyte.
• Demonstration of accuracy, where assay of standard reference materials is used to ensure analytical specificity and accurate quantification.
• Determination of the range, where the acceptable window of concentrations that adhere to the above steps is determined.
• Widening the scope by testing different parameters to demonstrate method suitability and reproducibility in other laboratories.
• Establishing the limits of detection and quantification, where the typical 3σ and 10σ criteria are used to assess sensitivity. In addition, the method quantitation limit, which accounts for all potential sources of error, from sample receipt to interpretation of final results.
• Establishing stability to ensure consistent results are reported over time.
• Assessing robustness, where small changes to experimental conditions are tested to assess if analytical validity is maintained.
Though this provides a useful guide, there is no international standard for method validation in clinical chemistry. Instead, the guidelines set in the ‘FDA Guidance for Industry – Bioanalytical Method Validation’112 used in the USA are the current benchmark worldwide. One significant limitation is the lack of appropriate standard reference materials, a key component to complete method validation. Well characterised mixtures of protein standards are available,113 though these encompass only a fraction of the human proteome. These materials are still particularly useful in method development studies and are supplemented by the freely-accessible and growing databases, such as the Human Proteome Organisation's Plasma Proteome Project, which was a multi-laboratory endeavor to fully characterise a pooled plasma sample.114 Additionally, this project highlights the importance of standardised analytical methods, from sample preparation though to analysis and examination of data, in ensuring reproducibility between sites. Theodorsson's115 extensive overview of method validation in clinical chemistry cites the analytical rigor to which the pharmaceutical industry is subjected as being the major driver of analytical validity in the clinical laboratory. International standards, such as ISO/IEC 17025 (General requirements for the competence of testing and calibration laboratories) and ISO 15189 (Medical laboratories—Particular requirements for quality and competence), provide laboratories with accreditation that ensures a standard of measurement is maintained.
However, better understanding of biochemistry highlights some concerns with many commonly used clinical tests. For instance, the established method used for assessing both ceruloplasmin and non-ceruloplasmin bound copper in serum uses a simple mathematical formula based on the assumption that ceruloplasmin is the sole copper-binding species. This was recently revisited by Twomey et al.,116 who found that confounding factors, such as copper non-specifically finding to other serum proteins, could result in incorrect interpretation of results. Similarly, use of LC-ICP-MS has been used to show that traditional immunoturbidity methods used for assessing transferrin saturation with iron are also limited by similar effects and are not sensitive to small variation.117 We raise the question that some established biochemical assays currently in use in the pathology laboratory could be revisited with the intent of improving analytical validity. Additionally, it is important to understand that the diversity of tissue types found in the human body may preclude an existing clinical test from providing truly accurate results, which in turn may require extensive re-validation, or at least confirmation against an appropriate matrix-matched standard.
Sample preparation is also key to ensuring analytically valid results are obtained. This step in the analytical workflow has been, and remains, the most significant window for the introduction of error through sample loss and contamination. Significant effort has been made to reduce possibility of human error by integrating previously arduous tasks into automated workflows, employing robotics for typical sample extraction, preconcentration and analysis.118 Such approaches have a vibrant future in translation to the clinical, where robust and reproducible analytical workflows, from sample receipt to data analysis, are critical for standardised and transferable methods. The development of new analytical methods, such as those used in ‘omics’ sciences has also driven further refinement of existing sample preparation strategies,119 as well as providing opportunities for innovative new approaches (e.g. nanotechnology120). Additionally, manual handling of samples increases risk of exposure to both hazardous biological and chemical materials. Gałuszka et al.121 recently introduced 12 key principles of ‘green’ analytical chemistry, with the intent to reduce risks and hazards, in addition to minimising the environmental impact of waste products and excessive energy use.
Validation of analytical methods for the clinic is still ongoing, and will continue to do so as new strategies begin the long path of translational science. Through to 2009, the US Food and Drug Administration (FDA) had approved 209 quantitative assays for plasma-based proteins, accounting for barely 1% of the human proteome.122 Liotta et al.123 speculated in 2003 that the future of blood-based proteomics was the integration of “new technology created at the intersection of the fields of artificial intelligence, nanotechnology and proteomics” to “provide ‘nanoharvesting’ agents designed specifically to capture and amplify classes of [low molecular weight] biomarkers”; though in 2016 such a resource still seems some distance away. This is not to say, however, that progress is not being made. With the advent of technology that can identify small variations in incredibly complex mixtures, like the human proteome, comes challenges for biomarker validation. Rifai and colleagues124 noted that the biggest barrier for the translation of biomarkers discovered through proteomics is a misalignment between marker discovery and traditional analytical method validation. Petricoin et al.'s125 comprehensive 2002 vision for translating proteomics from “benchside promise into bedside reality” highlights how far the field has to come before what analytical chemists may consider routine assays in the academic world are realised in not only the medical research field, but also in the diagnosis and treatment of disease, though we optimistically predict that narrowing the gap between the two disciplines will see significant advances made in the next decade.
A vision for the future
A challenge in the translation of chemical techniques in the biosciences is to ensure that the most promising and valuable newly developed protocols are recognised and adopted by biological researchers. In order to ensure that both new technologies and reagents reach mainstream research use, there is a need for dissemination of these offerings, which is best achieved through commercialisation. Without such progress, chemical research activity will continue fruitlessly for many years without any contribution to biological studies. For example, time-gated luminescence microscopy has, for a number of years, promised to enable completely background-free imaging.126 In this field, much chemical research has focussed on development of luminescent probes (typically organometallic transition metal127,128 or lanthanoid complexes129,130) with long luminescence lifetimes, but in this field, subsequent biological studies are primarily employed using conventional (non-gated) fluorescence or confocal microscopy. Notable exceptions arise from research groups that have built their own time-gated microscopes,131,132 but no research has crossed from development to application stage. However, with recent involvement of industry in the provision of parts to build such microscopes,133 there may be greater uptake of such technology in the future.It is clear, therefore, that the development of reagents (such as fluorescent probes) for bioanalysis should focus most attention on existing and embedded technologies. At the same time, however, it is essential that such research keep abreast of advances in the available and widely used technologies. For example, confocal microscopes have superseded fluorescence microscopes in the majority of research institutes, and current confocal microscopes offer very high sensitivity. As a result, quantum yields as low as 1% can be routinely imaged very clearly, with even lower values accessible through more sophisticated techniques.134 Amongst chemists developing fluorescent probes, however, the mantra persists that quantum yields must be 10% or higher,135 an artefact of requirements of early fluorescence microscopes. Furthermore, there have been exciting advances in microscope technology: linear unmixing protocols enable separation of two concurrently-used fluorophores with similar emission colours,136 while time-resolved microscopes offer an additional dimension for the development of sensors, which remains largely unexploited.137
The challenge of widespread adoption of analytical methods for the biosciences is particularly clear in the provision of reagents, such as fluorescent sensors. Primarily due to a preference for a tried-and-true method over a novel technique, biological researchers will tend to use a well-established reagent, rather than a novel chemical that has not seen widespread use, despite any evident advantages. This is well illustrated in the use of calcein, and its cell permeable profluorescent analogue, calcein AM, to study the labile iron pool. Calcein was first reported as a sensor of calcium,138 and subsequently for use in cell viability assays,139 but since it was first reported as a sensor of iron in 1995,140 the use of calcein AM as a measure of iron levels has been reported countless times. Given its other applications, the probe clearly lacks selectivity, responding in fact to all transition metals. Furthermore, it operates in a turn-off fashion, whereby the presence of iron is signalled by a quenching of fluorescence. This means that in live cell imaging studies, the absence of probe is indistinguishable from the presence of iron. The commonly used workaround, involving subsequent addition of an iron chelator to determine the amount of probe, is an endpoint protocol, precluding use in temporal studies of cells. However, despite these drawbacks, and a number of elegant fluorescent iron sensors that are selective for iron and operate by turn-on fluorescence changes,141,142 calcein AM remains the reagent of choice for biological studies.
Poor utilisation of new reagents in biological studies is painfully clear more broadly in the field of probe development. While the field of probe design is prolific, there is a lack of translation from chemist to biologist. We recently conducted a survey of the fluorescent sensors for intracellular Cu(I),143 and found that of the eleven small molecule probes with demonstrated biological utility, only two had been used in subsequent investigations of copper biology, by the original authors or others, and only one had been used more than once. The outliers in this set are the copper sensor probes CS1144 and CS3145 from the Chang group. CS1 has been used in seven subsequent studies in systems ranging from bacteria to Alzheimer's models,146–152 while CS3 has been used in two studies.153,154 As for new technologies, lack of commercial availability for reagents is certainly a factor; in fact, the authors of one study of copper in neuroblastoma noted the lack of any commercial selective copper sensor as the reason for their choice of non-selective, but commercially available metal sensor to report on copper changes.155
Much like standardisation is needed for translational to the clinic, the push for researchers to develop new and innovative analytical methods results in highly specialised and often unachievable in alternative laboratories in the immediate period following publication. Take, for example, the impressive protocol for highly multiplexed imaging of breast tumour sections described by Giesen et al.156 Here, the authors made use of the CyTOF,157 a heavily modified ICP-MS using a time-of-flight mass analyser for simultaneous detection of transient signals, which is beyond the capacity of the sequential quadrupole-based analyser in 95+% of ICP-MS systems installed worldwide.47 Marketed as a ‘mass cytometer’,158 the CyTOF uses prepared reagent kits employing monoisotopic lanthanide markers for labelling proteins and biomolecules, which can then be analysed in solution and undergo sophisticated multivariate statistical analysis.159 First commercialised in 2009 and now in its third generation design, uptake of the CyTOF in the biosciences (29 papers recorded in PubMed to 2015) is a good example of an analytical technique designed specifically with biological applications in mind. Charlotte Giesen's impressive report took this idea even further, partnering herself and Detlef Günther, two independently noteworthy names in the analytical sciences, with leading cancer researchers to apply laser ablation imaging protocols to the CyTOF system with a pertinent biological question to answer. No longer restricted by the fluorescence cross-talk experienced using traditional microscopy, and with ground-breaking ablation cell design mentioned previously (see Wang et al.95), the team managed to simultaneously image 32 proteins and protein modifications at subcellular (1 μm) resolution. As the CyTOF is mass-dependent, expression levels of the labelled proteins could be deduced from signal intensity, allowing multivariate ‘SPADE’ (see Qiu et al.160) analysis to identify heterogeneity of major proteins in a set of 22 tumour samples. As revolutionary as this example is, and it certainly has much potential in a range of chronic disease scenarios, no further papers have since used this technology, and the release of a commercialised system (first alluded to by the CyTOF manufacturer Fluidigm in mid-2014161) was still “…in the near future” one year later.162
Duplication of methods from system to system is no more apparent than in the most expensive category of analytical technology: the synchrotron. There are around 40 operational facilities worldwide, ranging in energy output significantly depending on the design. The three ‘third generation’ synchrotrons (Advanced Photon Source in the USA; the European Synchrotron Radiation Facility in France, and SPring-8 in Japan) dwarf the others in terms of size and energy capacity; all three have storage rings able to maintain electrons at >6 GeV. This does not, however, mean other facilities are inferior to their larger cousins—rather, new facilities are often designed in consultation with users and the requirements of the research environment within which they are situated. Consequently, and even though the relatively small size of the synchrotron research community makes it one of the most collaborative, there are significant differences in design and capabilities from facility to facility. Though we discuss XFM here as a main example, it is important to recognise that imaging metals is just one capability of a synchrotron; the newly commissioned National Synchrotron Light Source (NSLS) 2 at Brookhaven National Laboratories in the USA will eventually house over 60 beamlines, each with its own unique purpose.
As we previously discussed in our Tutorial Review on the subject,86 detection of analytes by XFM is as reliant on the detector as it is the sample. Many beamlines employ highly sensitive silicon drift detectors, which have the advantage of providing very low limits of detection and are commercially available but are limited by slow detector overheads; whilst others, such as the Australian Synchrotron, PETRA in Germany and the NSLS2 have opted to use fast-scanning capabilities of the multi-channel Maia detector,163 which sacrifices some of the advantages of drift detectors for the benefit of high throughput analysis. Thus, translating an experiment performed on the Maia to a beamline where the detector not only operates in a different manner, but may also be positioned in a different geometry, requires close consultation with beamline support scientists. Fortunately, as mentioned above, the close working relationship between synchrotron scientists worldwide, often through necessity, is a microcosm of the collaborative environment we envisage will become commonplace in the future as analytical chemists and biologists further recognise the benefits of applied research.
Improving communication is just one step, and sometimes just making biomedical researchers aware of new advances in analytical sciences can be a challenge. Giesen et al.'s156 ground-breaking work using LA-CyTOF technology was visible to the broader research community through publication in a leading biotechnology journal, yet it was made possible through years of work by scientists preceding them in the field of method development. Unfortunately, resources for medical and biological researchers such as PubMed often do not index analytical development journals, though publishers specialising in the chemical sciences have done a good job of: (1) recognising the importance of interface science when selecting papers to be reviewed; and (2) having their relevant journals indexed in PubMed as soon as is practicable. Social media also has a place in cross-disciplinary communication: a 2007 study reported that 77% of life scientists participate in some form of online social media,164 a number that has undoubtedly grown since these statistics were recorded. Every journal has an online presence, and with that comes all the necessary social media outlets, such as Twitter. However, it is not surprising that of the 20 top scientific personalities on Twitter (all with professional training in their discipline, though several have crossed from being active researchers to media entities),165 there is not one analytical chemist. Furthermore, it also disappointing that no female scientist makes this list, nor are there many who would be considered young researchers.
Concluding remarks: the analytical chemist of the future
As young investigators looking forward to careers in this field, it is timely to consider, then, what a chemist working in this space should look like. While the traditional model for this field has been for chemists to work on technological or reagent development, relatively independently of those researchers using these tools in detailed biological studies. It is clear, however, that this is not the way forward, and that chemists must now have a working knowledge of the biological questions they are seeking to probe, and ideally should have ready access to simple set-ups to perform preliminary biological validation studies. However, we must keep in mind that scientific progress relies on collaborations between experts with a depth of knowledge in one field rather than a shallow breadth, and it is therefore also essential analytical chemists of the future foster a diverse range of collaborations with various biological researchers. Collaborators will be able to provide advice on targets to be sensed, and can test the utility and generalisability of new tools developed.Beyond the broad generalisations of the breadth of expertise required, the analytical chemist of the future must strive to answer important biological questions, rather than seeking easy targets (the ‘low hanging fruit’ paradigm) that may have less value in biological studies. At the same time, it is essential that researchers are able to identify when application of developed methods is preferable to seeking to make improvements to technology. For the chemist, the tweaking of instruments, protocols or reagents can be facile, and while this can facilitate more rapid publication rates, such small changes are likely to have little impact in the broader scientific field. In contrast, true impact of new tools will be evident through their widespread application, so the exercise of validating and demonstrating the utility of a single tool in a broad range of studies will have greater impact.
The analytical chemist of the future must also be ambitious. The most exciting research will lie not in measuring analytes that have already been well studied, but in identifying prevailing challenges in biology for which there are no current analytical tools available. Furthermore, an analytical chemist must be a salesperson; promoting not only existing tools, but expertise to develop new tools in the future to as broad an audience as possible. It is only through cross-disciplinary discussion that the enormous potential of analytical chemistry can be harnessed.
It is helpful for emerging researchers to identify role models, and in this field, none is more appropriate than Roger Tsien. Perhaps best known for sharing the Nobel Prize in Chemistry in 2008 “for the discovery and development of the green fluorescent protein, GFP” (Tsien's team first described GFP's crystal structure in 1996166), his work and legacy has extended far beyond this field. As an emerging investigator himself, Tsien, at the time completing a PhD in neurophysiology in the late 1970s, used his expertise in chemistry to identify a key challenge for the area of neurochemistry: the ability to better visualise neuronal activity. He identified calcium as an appropriate analyte to study, and subsequently developed the first fluorescent sensor for calcium.167 This work pioneered the field of small molecule fluorescent sensors, and his innovation alone demonstrates the profound impact of such tools: calcium sensors are now routinely used to study neuronal activity. Over thirty years ago, Tsien was truly ahead of his time, and he exemplifies our description of the analytical chemist of the future, a model that is disappointingly rare amongst the current generation of researchers. Tsien's development of analytical tools was spurred directly by a biological question, and having produced a tool to address this need, he subsequently demonstrated its use in various biological studies. He also continued on, pioneering the use of ratiometric probes that signal analytes by a change in colour rather than intensity,168 and sensors for numerous other classes of analytes.169–171
It is certainly an exciting time to be an analytical chemist working towards a better understanding of the biosciences. It is encouraging that more biological research laboratories are actively integrating dedicated analytical chemistry development groups into the wider research teams. These are often centred on the concept of life sciences ‘innovative ecosystems’,172 where conglomerations of dedicated research institutes are geographically positioned within centralised locations, such as those found in Boston in the United States, Cambridge in the United Kingdom and Melbourne in Australia. The concept of innovation ecosystems, explained in the wider economic sense by Enrico Moretti,173 has the potential to build ‘brain hubs’, where multidisciplinary scientists are encouraged to share ideas and promote both commercial (in the case of industry-funded or university-sponsored innovation ecosystems) and academic innovation. Though typically viewed as a pipeline for commercialisation of research, these hubs of academic endeavour also have the potential to produce greater cohesion between multiple disciplines, and all the intellectual benefit that comes with it. Though the capital investment is large, many countries are recognising the benefits of such a model, and it is a direction that should encourage more analytical chemists and biologists to engage in sustained dialogue, so that both respective fields continue to flourish in an age where innovation continues to breed the most exciting discoveries. Though the gulf between analytical development and application within the biosciences is still wide, trends like these are indicative of a positive shift towards a more unified approach to what we should all consider a common goal: the pursuit of knowledge.
Acknowledgements
We would like to thank Dr Simon James, Dr Ian Birchall and Mirjana Bogeski of the Florey Institute of Neuroscience and Mental Health for assistance with figure components. D. J. H. is supported by an Australian Research Council Linkage Project (LP140100095) with Agilent Technologies.Notes and references
- T. L. Brown, H. E. LeMay, B. E. Bursten, C. Murphy, P. Woodward and M. E. Stoltzfus, Chemistry: The Central Science, Prentice Hall, 13th edn, 2014 Search PubMed.
- L. Cong, F. A. Ran, D. Cox, S. Lin, R. Barretto, N. Habib, P. D. Hsu, X. Wu, W. Jiang, L. A. Marraffini and F. Zhang, Science, 2013, 339, 819–823 CrossRef CAS PubMed.
- K. Chung, J. Wallace, S.-Y. Kim, S. Kalyanasundaram, A. S. Andalman, T. J. Davidson, J. J. Mirzabekov, K. A. Zalocusky, J. Mattis, A. K. Denisin, S. Pak, H. Bernstein, C. Ramakrishnan, L. Grosenick, V. Gradinaru and K. Deisseroth, Nature, 2013, 497, 332–337 CrossRef CAS PubMed.
- T. Walzthoeni, A. Leitner, F. Stengel and R. Aebersold, Curr. Opin. Struct. Biol., 2013, 23, 252–260 CrossRef CAS PubMed.
- S. J. Opella and F. M. Marassi, Chem. Rev., 2004, 104, 3587–3606 CrossRef CAS PubMed.
- E. J. New, Dalton Trans., 2013, 42, 3210–3219 RSC.
- R. Van Noorden, B. Maher and R. Nuzzo, Nature, 2014, 514, 550–553 CrossRef CAS PubMed.
- O. Lowry, N. Rosebrough, A. Farr and R. Randall, J. Biol. Chem., 1951, 193, 265–275 CAS.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- J. A. DiMasi, R. W. Hansen and H. G. Grabowski, J. Health Econ., 2003, 22, 151–185 CrossRef PubMed.
- I. W. Griffiths, Rapid Commun. Mass Spectrom., 1997, 11, 2–16 CrossRef CAS.
- T. Prohaska, in Sector Field Mass Spectrometry for Elemental and Isotopic Analysis, ed. T. Prohaska, J. Irrgeher, A. Zitek and N. Jakubowski, Royal Society of Chemistry, 2014, pp. 10–25 Search PubMed.
- D. Evanko, A. Heinrichs and C. K. Rosenthal, Nature Milestones: Light Microscopy, Nature Publishing Group, 2009 Search PubMed.
- A. Doerr, J. Finkelstein, I. Jarchum, C. Goodman and B. Dekker, Nature Milestones: Mass Spectrometry, Nature Publishing Group, 2015 Search PubMed.
- F. Szabadváry, in History of Analytical Chemistry, ed. F. Szabadváry, Pergamon, 1966, vol. 26, pp. 85–113 Search PubMed.
- K. H. Koch, Process Analytical Chemistry: Control, Optimization, Quality, Economy, Springer, 2013 Search PubMed.
- A. Pollard, C. Batt, B. Stern and S. Young, Analytical Chemistry in Archaeology, Cambridge University Press, 2007 Search PubMed.
- D. Pérez-Bendito and S. Rubio, Environmental Analytical Chemistry, Elsevier, 1999 Search PubMed.
- M. R. Siddiqui, Z. A. AlOthman and N. Rahman, Arabian J. Chem., 2013 DOI:10.1016/j.arabjc.2013.04.016.
- S. Hanson and T. Overton, Skills required by new chemistry graduates and their development in degree programmes, Higher Education Academy UK Physical Sciences Centre, Hull, UK, 2010 Search PubMed.
- R. Salzer, P. Taylor, N. H. Majcen, F. De Angelis, S. Wilmet, E. Varella and I. Kozaris, Chem. – Eur. J., 2015, 21, 9921–9935 CrossRef CAS PubMed.
- G. Gōmōri, Am. J. Pathol., 1936, 12, 655–664 Search PubMed.
- G. Orchard and B. Nation, Histopathology, Oxford University Press, Oxford, 2011 Search PubMed.
- D. P. Perl and P. F. Good, Ann. Neurol., 1992, 32, S76–S81 CrossRef CAS PubMed.
- G. A. Evans, Nat. Biotechnol., 2000, 18, 127 CrossRef CAS PubMed.
- P. James, Q. Rev. Biophys., 1997, 30, 279–331 CrossRef CAS PubMed.
- M. R. Wilkins, C. Pasquali, R. D. Appel, K. Ou, O. Golaz, J. C. Sanchez, J. X. Yan, A. A. Gooley, G. Hughes, I. Humphery-Smith, K. L. Williams and D. F. Hochstrasser, Nat. Biotechnol., 1996, 14, 61–65 CrossRef CAS.
- A. Mortazavi, B. A. Williams, K. McCue, L. Schaeffer and B. Wold, Nat. Methods, 2008, 5, 621–628 CrossRef CAS PubMed.
- S. G. Oliver, M. K. Winson, D. B. Kell and F. Baganz, Trends Biotechnol., 1998, 16, 373–379 CrossRef CAS PubMed.
- L. Kiemer and G. Cesareni, Trends Biotechnol., 2007, 25, 448–454 CrossRef CAS PubMed.
- H. Haraguchi, J. Anal. At. Spectrom., 2004, 19, 5 RSC.
- K. J. Waldron, J. C. Rutherford, D. Ford and N. J. Robinson, Nature, 2009, 460, 823–830 CrossRef CAS PubMed.
- A. Lothian, D. J. Hare, R. Grimm, T. M. Ryan, C. L. Masters and B. R. Roberts, Front. Aging Neurosci., 2013, 5, 35 Search PubMed.
- A. Cvetkovic, A. L. Menon, M. P. Thorgersen, J. W. Scott, F. L. Poole II, F. E. Jenney Jr, W. A. Lancaster, J. L. Praissman, S. Shanmukh, B. J. Vaccaro, S. A. Trauger, E. Kalisiak, J. V. Apon, G. Siuzdak, S. M. Yannone, J. A. Tainer and M. W. W. Adams, Nature, 2010, 466, 779–782 CrossRef CAS PubMed.
- F. Sanger, S. Nicklen and A. R. Coulson, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 5463–5467 CrossRef CAS.
- E. S. Lander, L. M. Linton, B. Birren, C. Nusbaum, M. C. Zody, J. Baldwin, K. Devon, K. Dewar, M. Doyle, W. FitzHugh, R. Funke, D. Gage, K. Harris, A. Heaford, J. Howland, L. Kann, J. Lehoczky, R. LeVine, P. McEwan, K. McKernan, J. Meldrim, J. P. Mesirov, C. Miranda, W. Morris, J. Naylor, C. Raymond, M. Rosetti, R. Santos, A. Sheridan, C. Sougnez, N. Stange-Thomann, N. Stojanovic, A. Subramanian, D. Wyman, J. Rogers, J. Sulston, R. Ainscough, S. Beck, D. Bentley, J. Burton, C. Clee, N. Carter, A. Coulson, R. Deadman, P. Deloukas, A. Dunham, I. Dunham, R. Durbin, L. French, D. Grafham, S. Gregory, T. Hubbard, S. Humphray, A. Hunt, M. Jones, C. Lloyd, A. McMurray, L. Matthews, S. Mercer, S. Milne, J. C. Mullikin, A. Mungall, R. Plumb, M. Ross, R. Shownkeen, S. Sims, R. H. Waterston, R. K. Wilson, L. W. Hillier, J. D. McPherson, M. A. Marra, E. R. Mardis, L. A. Fulton, A. T. Chinwalla, K. H. Pepin, W. R. Gish, S. L. Chissoe, M. C. Wendl, K. D. Delehaunty, T. L. Miner, A. Delehaunty, J. B. Kramer, L. L. Cook, R. S. Fulton, D. L. Johnson, P. J. Minx, S. W. Clifton, T. Hawkins, E. Branscomb, P. Predki, P. Richardson, S. Wenning, T. Slezak, N. Doggett, J.-F. Cheng, A. Olsen, S. Lucas, C. Elkin, E. Uberbacher, M. Frazier, R. A. Gibbs, D. M. Muzny, S. E. Scherer, J. B. Bouck, E. J. Sodergren, K. C. Worley, C. M. Rives, J. H. Gorrell, M. L. Metzker, S. L. Naylor, R. S. Kucherlapati, D. L. Nelson, G. M. Weinstock, Y. Sakaki, A. Fujiyama, M. Hattori, T. Yada, A. Toyoda, T. Itoh, C. Kawagoe, H. Watanabe, Y. Totoki, T. Taylor, J. Weissenbach, R. Heilig, W. Saurin, F. Artiguenave, P. Brottier, T. Bruls, E. Pelletier, C. Robert, P. Wincker, A. Rosenthal, M. Platzer, G. Nyakatura, S. Taudien, A. Rump, D. R. Smith, L. Doucette-Stamm, M. Rubenfield, K. Weinstock, H. M. Lee, J. Dubois, H. Yang, J. Yu, J. Wang, G. Huang, J. Gu, L. Hood, L. Rowen, A. Madan, S. Qin, R. W. Davis, N. A. Federspiel, A. P. Abola, M. J. Proctor, B. A. Roe, F. Chen, H. Pan, J. Ramser, H. Lehrach, R. Reinhardt, W. R. McCombie, M. de la Bastide, N. Dedhia, H. Blöcker, K. Hornischer, G. Nordsiek, R. Agarwala, L. Aravind, J. A. Bailey, A. Bateman, S. Batzoglou, E. Birney, P. Bork, D. G. Brown, C. B. Burge, L. Cerutti, H.-C. Chen, D. Church, M. Clamp, R. R. Copley, T. Doerks, S. R. Eddy, E. E. Eichler, T. S. Furey, J. Galagan, J. G. R. Gilbert, C. Harmon, Y. Hayashizaki, D. Haussler, H. Hermjakob, K. Hokamp, W. Jang, L. S. Johnson, T. A. Jones, S. Kasif, A. Kaspryzk, S. Kennedy, W. J. Kent, P. Kitts, E. V. Koonin, I. Korf, D. Kulp, D. Lancet, T. M. Lowe, A. McLysaght, T. Mikkelsen, J. V. Moran, N. Mulder, V. J. Pollara, C. P. Ponting, G. Schuler, J. Schultz, G. Slater, A. F. A. Smit, E. Stupka, J. Szustakowki, D. Thierry-Mieg, J. Thierry-Mieg, L. Wagner, J. Wallis, R. Wheeler, A. Williams, Y. I. Wolf, K. H. Wolfe, S.-P. Yang, R.-F. Yeh, F. Collins, M. S. Guyer, J. Peterson, A. Felsenfeld, K. A. Wetterstrand, R. M. Myers, J. Schmutz, M. Dickson, J. Grimwood, D. R. Cox, M. V. Olson, R. Kaul, C. Raymond, N. Shimizu, K. Kawasaki, S. Minoshima, G. A. Evans, M. Athanasiou, R. Schultz, A. Patrinos and M. J. Morgan, Nature, 2001, 409, 860–921 CrossRef CAS PubMed.
- Z. Wang, M. Gerstein and M. Snyder, Nat. Rev. Genet., 2009, 10, 57–63 CrossRef CAS PubMed.
- D. R. Smith, Front. Genet., 2013, 4, 72 Search PubMed.
- S. Pabinger, A. Dander, M. Fischer, R. Snajder, M. Sperk, M. Efremova, B. Krabichler, M. R. Speicher, J. Zschocke and Z. Trajanoski, Briefings Bioinf., 2014, 15, 256–278 CrossRef PubMed.
- N. J. Loman, C. Constantinidou, J. Z. M. Chan, M. Halachev, M. Sergeant, C. W. Penn, E. R. Robinson and M. J. Pallen, Nat. Rev. Microbiol., 2012, 10, 599–606 CrossRef CAS PubMed.
- The Impact of Genomics on the US Economy, Battelle, 2013.
- T. Rabilloud, Proteomics, 2002, 2, 3–10 CrossRef CAS PubMed.
- S. Magdeldin, S. Enany, Y. Yoshida, B. Xu, Y. Zhang, Z. Zureena, I. Lokamani, E. Yaoita and T. Yamamoto, Clin. Proteomics, 2014, 11, 16 CrossRef PubMed.
- X. He, Q. Chen, Y. Zhang and J.-M. Lin, TrAC, Trends Anal. Chem., 2014, 53, 84–97 CrossRef CAS.
- K. K. Unger, K. Racaityte, K. Wagner, T. Miliotis, L. E. Edholm, R. Bischoff and G. Marko Varga, J. High Resolut. Chromatogr., 2000, 23, 259–265 CrossRef CAS.
- P. Pomastowski and B. Buszewski, TrAC, Trends Anal. Chem., 2014, 53, 167–177 CrossRef CAS.
- D. Potter, J. Anal. At. Spectrom., 2008, 23, 690 RSC.
- K. Nier, A. Yergey and P. Gale, The Encyclopedia of Mass Spectrometry: Volume 9: Historical Perspectives, Part B: Notable People in Mass Spectrometry, Elsevier, 2015 Search PubMed.
- S. Eliuk and A. Makarov, Annu. Rev. Anal. Chem., 2015, 8, 61–80 CrossRef PubMed.
- S. Diez Fernández, N. Sugishama, J. Ruiz Encinar and A. Sanz-Medel, Anal. Chem., 2012, 84, 5851–5857 CrossRef PubMed.
- L. Balcaen, E. Bolea-Fernandez, M. Resano and F. Vanhaecke, Anal. Chim. Acta, 2015, 894, 7–19 CrossRef CAS PubMed.
- R. M. Caprioli, T. B. Farmer and J. Gile, Anal. Chem., 1997, 69, 4751–4760 CrossRef CAS PubMed.
- D. S. Cornett, M. L. Reyzer, P. Chaurand and R. M. Caprioli, Nat. Methods, 2007, 4, 828–833 CrossRef CAS PubMed.
- M. Aichler and A. Walch, Lab. Invest., 2015, 95, 422–431 CrossRef CAS PubMed.
- S. Maher, F. P. M. Jjunju and S. Taylor, Rev. Mod. Phys., 2015, 87, 113–135 CrossRef.
- D. Kell, Curr. Opin. Microbiol., 2004, 7, 296–307 CrossRef CAS PubMed.
- W. E. Steiner and W. A. English, J. Anal. Bioanal. Tech., 2015, 6, 1–3 Search PubMed.
- O. Quehenberger, A. M. Armando and E. A. Dennis, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2011, 1811, 648–656 CrossRef CAS PubMed.
- M. Vinaixa, E. L. Schymanski, S. Neumann, M. Navarro, R. M. Salek and O. Yanes, TrAC, Trends Anal. Chem., 2016, 78, 23–35 CrossRef CAS.
- S. Pal, B. Sen, S. Lohar, M. Mukherjee, S. Banerjee and P. Chattopadhyay, Dalton Trans., 2015, 44, 1761–1768 RSC.
- D. Montagner, S. Q. Yap and W. H. Ang, Angew. Chem., Int. Ed., 2013, 52, 11785–11789 CrossRef CAS PubMed.
- C. Shen, B. D. W. Harris, L. J. Dawson, K. A. Charles, T. W. Hambley and E. J. New, Chem. Commun., 2015, 51, 6312–6314 RSC.
- K. P. Carter, A. M. Young and A. E. Palmer, Chem. Rev., 2014, 114, 4564–4601 CrossRef CAS PubMed.
- X. Liu, N. Zhang, J. Zhou, T. Chang, C. Fang and D. Shangguan, Analyst, 2013, 138, 901–906 RSC.
- T. D. Ashton, K. A. Jolliffe and F. M. Pfeffer, Chem. Soc. Rev., 2015, 44, 4547–4595 RSC.
- H. Koga, S. Kotoh, M. Nakashima, A. Yokomizo, M. Tanaka and S. Naito, Int. J. Oncol., 2000, 16, 1003–1007 CAS.
- L. Yu, M. H. Chen, Z. J. Li, J. Wen, J. H. Fu, D. Guo, Y. Jiang, S. G. Wu, C. H. Cho and S. W. Liu, Mol. Pharmacol., 2011, 79, 608–617 CrossRef CAS PubMed.
- A. V. Klein and T. W. Hambley, Chem. Rev., 2009, 109, 4911–4920 CrossRef CAS PubMed.
- R. Meguro, Y. Asano, H. Iwatsuki and K. Shoumura, Histochem. Cell Biol., 2003, 120, 73–82 CrossRef CAS PubMed.
- R. Meguro, Y. Asano, S. Odagiri, C. Li, H. Iwatsuki and K. Shoumura, Arch. Histol. Cytol., 2007, 70, 1–19 CrossRef CAS PubMed.
- D. J. Hare, P. Lei, S. Ayton, B. R. Roberts, R. Grimm, J. L. George, D. P. Bishop, A. D. Beavis, S. J. Donovan, G. McColl, I. Volitakis, C. L. Masters, P. A. Adlard, R. A. Cherny, A. I. Bush, D. I. Finkelstein and P. A. Doble, Chem. Sci., 2014, 5, 2160–2169 RSC.
- D. J. Hare, C. Austin and P. Doble, Analyst, 2012, 137, 1527–1537 RSC.
- D. J. Hare, J. Lear, D. Bishop, A. Beavis and P. A. Doble, Anal. Methods, 2013, 5, 1915–1921 RSC.
- K. W. Dunn, M. M. Kamocka and J. H. McDonald, Am. J. Physiol.: Cell Physiol., 2011, 300, C723–C742 CrossRef CAS PubMed.
- D. J. Hare, B. Reedy, R. Grimm, S. Wilkins, I. Volitakis, J. L. George, R. A. Cherny, A. I. Bush, D. I. Finkelstein and P. Doble, Metallomics, 2009, 1, 53 RSC.
- D. P. Bishop, D. Clases, F. Fryer, E. Williams, S. Wilkins, D. J. Hare, N. Cole, U. Karst and P. A. Doble, J. Anal. At. Spectrom., 2016, 31, 197–202 RSC.
- D. J. Hare, M. W. M. Jones, V. C. Wimmer, N. L. Jenkins, M. D. de Jonge, A. I. Bush and G. McColl, Metallomics, 2016, 8, 156–160 RSC.
- A. Napolitano, A. Pezzella and G. Prota, Chem. Res. Toxicol., 1999, 12, 1090–1097 CrossRef CAS PubMed.
- K. Honarmand Ebrahimi, P.-L. Hagedoorn and W. R. Hagen, Chem. Rev., 2015, 115, 295–326 CrossRef CAS PubMed.
- L. Mercer, M. del Fiacco and A. C. Cuello, Experientia, 1979, 35, 101–103 CrossRef CAS PubMed.
- D. J. Hare, P. A. Adlard, P. A. Doble and D. I. Finkelstein, Metallomics, 2013, 5, 91–109 RSC.
- R. Wasserman, The Transfer of Calcium and Strontium Across Biological Membranes, Academic Press, New York, 2012 Search PubMed.
- P. Doble, M. Sandercock, E. Du Pasquier, P. Petocz, C. Roux and M. Dawson, Forensic Sci. Int., 2003, 132, 26–39 CrossRef CAS PubMed.
- J. J. Ramsden, Bioinformatics: An Introduction, Springer London, London, 2009, pp. 1–6 Search PubMed.
- L. Eriksson, H. Antti, J. Gottfries, E. Holmes, E. Johansson, F. Lindgren, I. Long, T. Lundstedt, J. Trygg and S. Wold, Anal. Bioanal. Chem., 2004, 380, 419–429 CrossRef CAS PubMed.
- D. J. Hare, E. J. New, M. D. de Jonge and G. McColl, Chem. Soc. Rev., 2015, 44, 5941–5958 RSC.
- B. Chen, S. Heng, H. Peng, B. Hu, X. Yu, Z. Zhang, D. Pang, X. Yue and Y. Zhu, J. Anal. At. Spectrom., 2010, 25, 1931–1938 RSC.
- C. J. Fahrni, Curr. Opin. Chem. Biol., 2007, 11, 121–127 CrossRef CAS PubMed.
- M. J. Hackett, J. A. McQuillan, F. El-Assaad, J. B. Aitken, A. Levina, D. D. Cohen, R. Siegele, E. A. Carter, G. E. Grau, N. H. Hunt and P. A. Lay, Analyst, 2011, 136, 2941–2952 RSC.
- S. Chen, J. Deng, Y. Yuan, C. Flachenecker, R. Mak, B. Hornberger, Q. Jin, D. Shu, B. Lai, J. Maser, C. Roehrig, T. Paunesku, S. C. Gleber, D. J. Vine, L. Finney, J. VonOsinski, M. Bolbat, I. Spink, Z. Chen, J. Steele, D. Trapp, J. Irwin, M. Feser, E. Snyder, K. Brister, C. Jacobsen, G. Woloschak and S. Vogt, J. Synchrotron Radiat., 2014, 21, 66–75 CAS.
- T. Ishizuka and Y. Uwamino, Spectrochim. Acta, Part B, 1983, 38, 519–527 CrossRef.
- A. L. Gray, Analyst, 1985, 110, 551–556 RSC.
- S. F. Durrant, J. Anal. At. Spectrom., 1999, 14, 1385–1403 RSC.
- S. Wang, R. Brown and D. J. Gray, Appl. Spectrosc., 1994, 48, 1321–1325 CrossRef CAS.
- H. A. O. Wang, D. Grolimund, C. Giesen, C. N. Borca, J. R. H. Shaw-Stewart, B. Bodenmiller and D. Günther, Anal. Chem., 2013, 85, 10107–10116 CrossRef CAS PubMed.
- C. Barillari, D. Ottoz, J. M. Fuentes-Serna, C. Ramakrishnan, B. Rinn and F. Rudolf, Bioinformatics, 2016, 32, 638–640 CrossRef PubMed.
- L. D. Lavis and R. T. Raines, ACS Chem. Biol., 2008, 3, 142–155 CrossRef CAS PubMed.
- G. K. Walkup, S. C. Burdette, S. J. Lippard and R. Y. Tsien, J. Am. Chem. Soc., 2000, 122, 5644–5645 CrossRef CAS.
- T. Hirano, K. Kikuchi, Y. Urano and T. Nagano, J. Am. Chem. Soc., 2002, 124, 6555–6562 CrossRef CAS PubMed.
- C. J. Chang, J. Jaworski, E. M. Nolan, M. Sheng and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 1129–1134 CrossRef CAS PubMed.
- W. Chyan, D. Y. Zhang, S. J. Lippard and R. J. Radford, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 143–148 CrossRef CAS PubMed.
- H. Sasaki, K. Hanaoka, Y. Urano, T. Terai and T. Nagano, Bioorg. Med. Chem., 2011, 19, 1072–1078 CrossRef CAS PubMed.
- R. J. Radford, W. Chyan and S. J. Lippard, Chem. Sci., 2014, 5, 4512–4516 RSC.
- T. Egawa, K. Hanaoka, Y. Koide, S. Ujita, N. Takahashi, Y. Ikegaya, N. Matsuki, T. Terai, T. Ueno, T. Komatsu and T. Nagano, J. Am. Chem. Soc., 2011, 133, 14157–14159 CrossRef CAS PubMed.
- A. M. Smith, M. C. Mancini and S. Nie, Nat. Nanotechnol., 2009, 4, 710–711 CrossRef CAS PubMed.
- S. L. Gibbs, Quant. Imaging Med. Surg., 2012, 2, 177–187 Search PubMed.
- F. G. Strathmann and A. N. Hoofnagle, Am. J. Clin. Pathol., 2011, 136, 609–616 CrossRef CAS PubMed.
- A. N. Hoofnagle, Clin. Chem., 2010, 56, 161–164 CAS.
- H. Mischak, R. Apweiler, R. E. Banks, M. Conaway, J. Coon, A. Dominiczak, J. H. H. Ehrich, D. Fliser, M. Girolami, H. Hermjakob, D. Hochstrasser, J. Jankowski, B. A. Julian, W. Kolch, Z. A. Massy, C. Neusuess, J. Novak, K. Peter, K. Rossing, J. Schanstra, O. J. Semmes, D. Theodorescu, V. Thongboonkerd, E. M. Weissinger, J. E. Van Eyk and T. Yamamoto, Proteomics: Clin. Appl., 2007, 1, 148–156 CrossRef CAS PubMed.
- M. Plebani, L. Sciacovelli, A. Aita, A. Padoan and M. L. Chiozza, Clin. Chim. Acta, 2014, 432, 44–48 CrossRef CAS PubMed.
- J. M. Green, Anal. Chem., 1996, 68, 305A–309A CrossRef CAS.
- FDA Guidance for Industry. Bioanalytical Method Validation (2001), http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm070107.pdf.
- A. R. Ivanov, C. M. Colangelo, C. P. Dufresne, D. B. Friedman, K. S. Lilley, K. Mechtler, B. S. Phinney, K. L. Rose, P. A. Rudnick, B. C. Searle, S. A. Shaffer and S. T. Weintraub, Proteomics, 2013, 13, 904–909 CrossRef CAS PubMed.
- G. S. Omenn, Clin. Chem. Lab. Med., 2004, 42, 1–2 CrossRef CAS PubMed.
- E. Theodorsson, Bioanalysis, 2012, 4, 305–320 CrossRef CAS PubMed.
- P. J. Twomey, A. Viljoen, I. M. House, T. M. Reynolds and A. S. Wierzbicki, Clin. Chem., 2005, 51, 1558–1559 CAS.
- D. J. Hare, J. D. Doecke, N. G. Faux, A. Rembach, I. Volitakis, C. J. Fowler, R. Grimm, P. A. Doble, R. A. Cherny, C. L. Masters, A. I. Bush and B. R. Roberts, ACS Chem. Neurosci., 2015, 6, 398–402 CrossRef CAS PubMed.
- N. Zheng, H. Jiang and J. Zeng, Bioanalysis, 2014, 6, 2441–2459 CrossRef CAS PubMed.
- D. Vuckovic, Anal. Bioanal. Chem., 2012, 403, 1523–1548 CrossRef CAS PubMed.
- Y. Li, X. Zhang and C. Deng, Chem. Soc. Rev., 2013, 42, 8517–8539 RSC.
- A. Gałuszka, Z. Migaszewski and J. Namieśnik, TrAC, Trends Anal. Chem., 2013, 50, 78–84 CrossRef.
- N. L. Anderson, Clin. Chem., 2010, 56, 177–185 CAS.
- L. A. Liotta, M. Ferrari and E. Petricoin, Nature, 2003, 425, 905 CrossRef CAS PubMed.
- N. Rifai, M. A. Gillette and S. A. Carr, Nat. Biotechnol., 2006, 24, 971–983 CrossRef CAS PubMed.
- E. F. Petricoin, K. C. Zoon, E. C. Kohn, J. C. Barrett and L. A. Liotta, Nat. Rev. Drug Discovery, 2002, 1, 683–695 CrossRef CAS PubMed.
- A. Beeby, S. W. Botchway, I. M. Clarkson, S. Faulkner, A. W. Parker, D. Parker and J. A. G. Williams, J. Photochem. Photobiol., B, 2000, 57, 83–89 CrossRef CAS.
- L. Murphy, A. Congreve, L. O. Palsson and J. A. G. Williams, Chem. Commun., 2010, 46, 8743–8745 RSC.
- C. A. Bader, R. D. Brooks, Y. S. Ng, A. Sorvina, M. V. Werrett, P. J. Wright, A. G. Anwer, D. A. Brooks, S. Stagni, S. Muzzioli, M. Silberstein, B. W. Skelton, E. M. Goldys, S. E. Plush, T. Shandala and M. Massi, RSC Adv., 2014, 4, 16345–16351 RSC.
- C. P. Montgomery, B. S. Murray, E. J. New, R. Pal and D. Parker, Acc. Chem. Res., 2009, 42, 925–937 CrossRef CAS PubMed.
- S. J. Butler, L. Lamarque, R. Pal and D. Parker, Chem. Sci., 2014, 5, 1750–1756 RSC.
- K. Hanaoka, K. Kikuchi, S. Kobayashi and T. Nagano, J. Am. Chem. Soc., 2007, 129, 13502–13509 CrossRef CAS PubMed.
- R. Pal and A. Beeby, Methods Appl. Fluoresc., 2014, 2, 3 Search PubMed.
- D. Jin, Y. Lu, R. C. Leif, S. Yang, M. Rajendran and L. W. Miller, Curr. Protoc. Cytom., 2014, 67, 2.22.1–2.22.36 Search PubMed.
- L. C. Estrada, M. J. Roberti, S. Simoncelli, V. Levi, P. F. Aramendía and O. E. Martínez, J. Phys. Chem. B, 2012, 116, 2306–2313 CrossRef CAS PubMed.
- D. Lansing Taylor and E. D. Salmon, in Methods in Cell Biology, Volume 29: Fluorescence Microscopy of Living Cells in Culture, Part A, ed. Y.-L. Wang and D. Lansing Taylor, Accademic Press, San Diego, California, 1989 Search PubMed.
- T. Zimmermann, in Microscopy Techniques, ed. J. Rietdorf, 2005, vol. 95, pp. 245–265 Search PubMed.
- K. Suhling, P. M. W. French and D. Phillips, Photochem. Photobiol. Sci., 2005, 4, 13–22 CAS.
- B. M. Tucker, Analyst, 1957, 82, 284–285 RSC.
- B. E. Frueh, M. Boehnke and J. Blumenstein, Invest. Ophthalmol. Visual Sci., 1993, 34, 771 Search PubMed.
- W. Breuer, S. Epsztejn and Z. I. Cabantchik, J. Biol. Chem., 1995, 270, 24209–24215 CrossRef CAS PubMed.
- L. M. Hyman and K. J. Franz, Coord. Chem. Rev., 2012, 256, 2333–2356 CrossRef CAS PubMed.
- H. Zhu, J. L. Fan, B. H. Wang and X. J. Peng, Chem. Soc. Rev., 2015, 44, 4337–4366 RSC.
- C. Shen and E. J. New, Metallomics, 2015, 7, 56–65 RSC.
- E. W. Miller, L. Zeng, D. W. Domaille and C. J. Chang, Nat. Protoc., 2006, 1, 824–827 CrossRef CAS PubMed.
- S. C. Dodani, D. W. Domaille, C. I. Nam, E. W. Miller, L. A. Finney, S. Vogt and C. J. Chang, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 5980–5985 CrossRef CAS PubMed.
- T. Miyayama, K. T. Suzuki and Y. Ogra, Toxicol. Appl. Pharmacol., 2009, 237, 205–213 CrossRef CAS PubMed.
- T. Miyayama, D. Hiraoka, F. Kawaji, E. Nakamura, N. Suzuki and Y. Ogra, Biochem. J., 2010, 429, 53–61 CrossRef CAS PubMed.
- M. Schrag, A. Crofton, M. Zabel, A. Jiffry, D. Kirsch, A. Dickson, X. W. Mao, H. V. Vinters, D. W. Domaille, C. J. Chang and W. Kirsch, J. Alzheimer's Dis., 2011, 24, 137–149 CAS.
- K. A. Price, J. L. Hickey, Z. G. Xiao, A. G. Wedd, S. A. James, J. R. Liddell, P. J. Crouch, A. R. White and P. S. Donnelly, Chem. Sci., 2012, 3, 2748–2759 RSC.
- H. W. Mbatia, H. M. D. Bandara and S. C. Burdette, Chem. Commun., 2012, 48, 5331–5333 RSC.
- K. D. Cusick, S. C. Minkin, S. C. Dodani, C. J. Chang, S. W. Wilhelm and G. S. Sayler, Environ. Sci. Technol., 2012, 46, 2959–2966 CrossRef CAS PubMed.
- J. Beaudoin, R. Ioannoni, L. López-Maury, J. Bähler, S. Ait-Mohand, B. Guérin, S. C. Dodani, C. J. Chang and S. Labbé, J. Biol. Chem., 2011, 286, 34356–34372 CrossRef CAS PubMed.
- C. P. Huang, M. Fofana, J. Chan, C. J. Chang and S. B. Howell, Metallomics, 2014, 6, 654–661 RSC.
- A. Hong-Hermesdorf, M. Miethke, S. D. Gallaher, J. Kropat, S. C. Dodani, J. Chan, D. Barupala, D. W. Domaille, D. I. Shirasaki, J. A. Loo, P. K. Weber, J. Pett-Ridge, T. L. Stemmler, C. J. Chang and S. S. Merchant, Nat. Chem. Biol., 2014, 10, 1034–1042 CrossRef CAS PubMed.
- E. Urso, A. Rizzello, R. Acierno, M. G. Lionetto, B. Salvato, C. Storelli and M. Maffia, J. Membr. Biol., 2010, 233, 13–21 CrossRef CAS PubMed.
- C. Giesen, H. A. O. Wang, D. Schapiro, N. Zivanovic, A. Jacobs, B. Hattendorf, P. J. Schüffler, D. Grolimund, J. M. Buhmann, S. Brandt, Z. Varga, P. J. Wild, D. Günther and B. Bodenmiller, Nat. Methods, 2014, 11, 417–422 CrossRef CAS PubMed.
- S. C. Bendall, E. F. Simonds, P. Qiu, E. A. D. Amir, P. O. Krutzik, R. Finck, R. V. Bruggner, R. Melamed, A. Trejo, O. I. Ornatsky, R. S. Balderas, S. K. Plevritis, K. Sachs, D. Pe'er, S. D. Tanner and G. P. Nolan, Science, 2011, 332, 687–696 CrossRef CAS PubMed.
- D. R. Bandura, V. I. Baranov, O. I. Ornatsky, A. Antonov, R. Kinach, X. Lou, S. Pavlov, S. Vorobiev, J. E. Dick and S. D. Tanner, Anal. Chem., 2009, 81, 6813–6822 CrossRef CAS PubMed.
- O. Ornatsky, D. Bandura, V. Baranov, M. Nitz, M. A. Winnik and S. Tanner, J. Immunol. Methods, 2010, 361, 1–20 CrossRef CAS PubMed.
- P. Qiu, E. F. Simonds, S. C. Bendall, K. D. Gibbs Jr, R. V. Bruggner, M. D. Linderman, K. Sachs, G. P. Nolan and S. K. Plevritis, Nat. Biotechnol., 2011, 29, 886–891 CrossRef CAS PubMed.
- Fluidigm Corporation, Press release: Early access program for new imaging mass cytometry platform, 31/6/2014.
- Fluidigm Corporation, Fluidigm Introduces Advanced Mass Cytometry System, The Scientist, 26/6/2015.
- C. G. Ryan, D. P. Siddons and G. Moorhead, J. Phys.: Conf. Ser., 2009, 186, 012013 CrossRef.
- L. Van Eperen and F. M. Marincola, J. Transl. Med., 2011, 9, 199 CrossRef PubMed.
- J. You, Science, 2014, 345, 1440–1441 CrossRef CAS PubMed.
- M. Ormö, A. B. Cubitt, K. Kallio, L. A. Gross, R. Y. Tsien and S. J. Remington, Science, 1996, 273, 1392–1395 Search PubMed.
- R. Y. Tsien, Biochemistry, 1980, 19, 2396–2404 CrossRef CAS PubMed.
- G. Grynkiewicz, M. Poenie and R. Y. Tsien, J. Biol. Chem., 1985, 260, 3440–3450 CAS.
- A. Minta and R. Y. Tsien, J. Biol. Chem., 1989, 264, 19449–19457 CAS.
- S. C. Burdette, G. K. Walkup, B. Spingler, R. Y. Tsien and S. J. Lippard, J. Am. Chem. Soc., 2001, 123, 7831–7841 CrossRef CAS PubMed.
- R. Weinstain, J. Kanter, B. Friedman, L. G. Ellies, M. E. Baker and R. Y. Tsien, Bioconjugate Chem., 2013, 24, 766–771 CrossRef CAS PubMed.
- K. D. Harrison, N. S. Kadaba, R. B. Kelly and D. Crawford, Sci. Transl. Med., 2012, 4, 157fs137 Search PubMed.
- E. Moretti, The New Geography of Jobs, Mariner Books, New York, 2013 Search PubMed.
Footnotes |
| † A variant of the Turnbull method to produce ferrous ferricyanide. |
| ‡ Turnbull's method with the addition of the reducing agent ammonium sulfide. |
| This journal is © The Royal Society of Chemistry 2016 |